
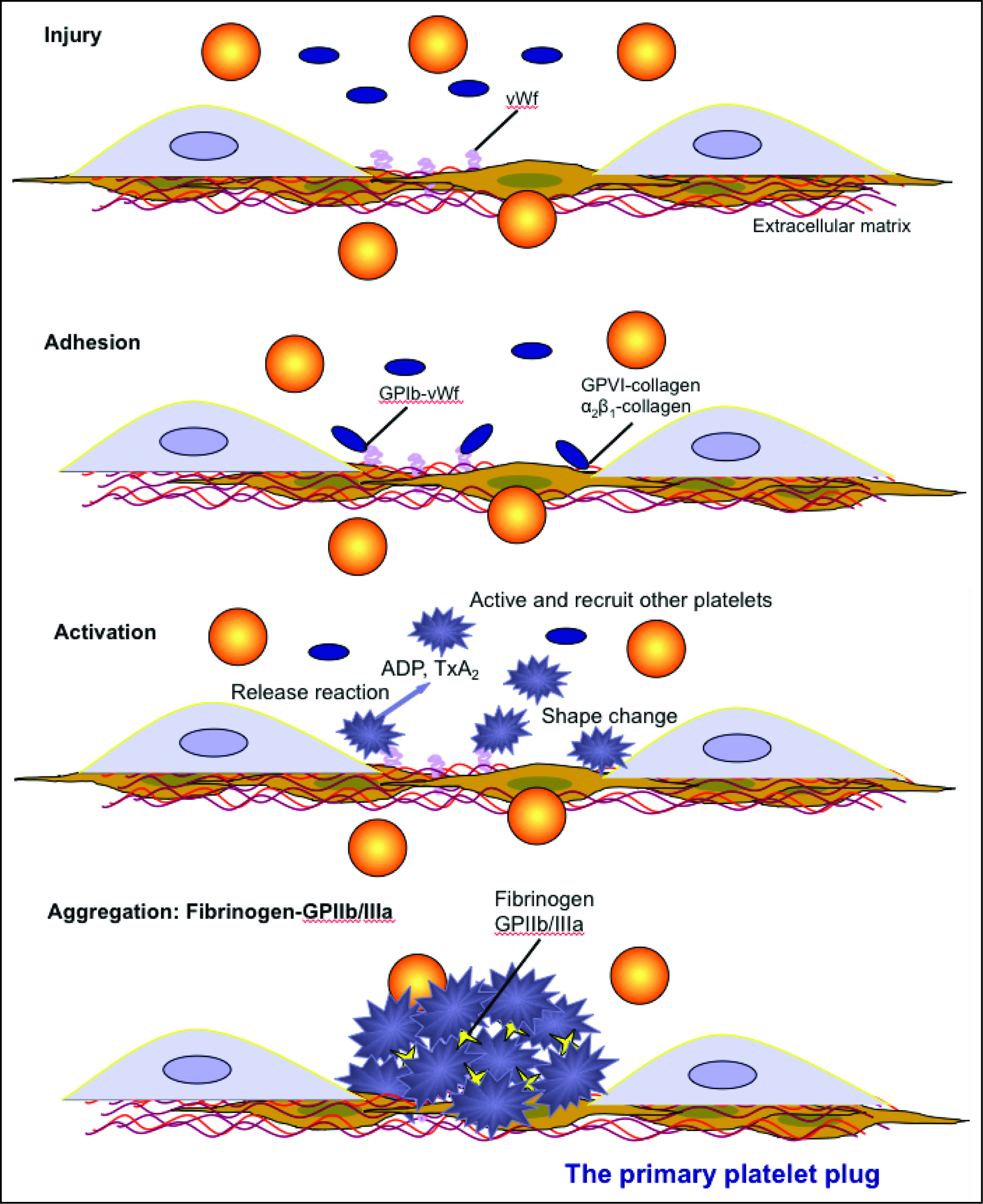
Endothelial injury exposes subendothelial matrix proteins (e.g. collagen) and von Willebrand factor (vWf) to circulating platelets. Platelets adhere to collagen (GPVI or the integrin α2β1 or vWf via surface receptors (GPIb-IX-V) and become activated through intracellular signaling. Once activated, platelets change shape, undergo phospholipid metabolism, which produces platelet agonists such as thromboxane A2 (TXA2) and release platelet agonists such as ADP, from dense granules. These agonists then activate and recruit unactivated circulating platelets. The fibrinogen receptor (GPIIb/IIIa or GPαIIbβ3) is activated by inside-out signaling, and binds fibrinogen, which bridges adjacent platelets forming a platelet plug. This is sufficient to stop hemorrhage in small blood vessels (e.g. mucosal vessels) but is insufficient in larger vessels or with severe injury. In the latter situations, the platelet plug must be stabilized via fibrin formation through secondary hemostasis. Platelets participate in secondary hemostasis (and fibrinolysis) by flipping their membranes exposing the anionic phospholipid, phosphatidylserine, which serves as a binding surface and amplification mechanism for coagulation factor complex assembly and activity. They also release phosphatidylserine-rich microparticles, which provide a large surface area on which fibrin formation proceeds. Short chain polyphosphates are also released from platelets (dense granules) as are stored coagulation factors such as factor V and factor XIII. This provides high local concentrations of these factors in the vicinity of the platelets, where fibrin formation is occurring. These factors can then facilitate fibrin formation (factor V is part of the “prothrombinase” and a cofactor for activated factor X on platelet surfaces, polyphosphates promote the cofactor activity of factor V, and factor XIII crosslinks soluble fibrin).