
Production
Erythrocytes are produced from hematopoietic stem cells in the bone marrow under the influence of the cytokine, erythropoietin, during steady state erythropoiesis. There is controversy as to the exact origin of the erythropoietin cell in the kidney. However, the main erythropoietin-producing cell in the kidney, particularly under states of hypoxia, is the peritubular interstitial cell, which is mostly found in the cortex near the junction of the cortex and medulla (Haase 2013, Shih et al 2018). There is also evidence in rats that erythropoietin is produced in renal tubular epithelial cells, particularly in intercalated cells in the collecting ducts, under steady state or normal conditions (Nagai et al 2014). In addition, low amounts of erythropoietin are produced in other sites including the liver (likely explaining secondary erythrocytosis due to some hepatic tumors, like hepatoblastoma [Lennox et al 2000, Axon et al 2008, Gold et al 2008]), testes, brain and spleen, however these sites cannot substitute for lack of renal production. The main stimulus for erythropoiesis is hypoxia or decreased tissue oxygen tension. This results in stabilization of the transcription factors, hypoxia-inducible factor (HIF), which normally are degraded under normoxic conditions (Shih et al 2018). HIF consists of two subunits, α and β and there are three different α variants (HIF-1α, HIF-2α, HIF-3α), with HIF-1α and HIF-2α having the main roles in erythropoiesis (Shih et al 2018). HIF bind to HIF-responsive elements (HRE) in the erythropoietin promoter, stimulating transcription (Souma et al 2015) and increasing erythropoietin production. It is currently thought that oxygen sensing and erythropoietin production occur in the same cell, ie. the peritubular or interstitial cell in states of hypoxia (and to a lesser extent tubular epithelial cells in rats (Nagai et al 2014)), but this has not been fully elucidated. Note that HIF expression is not restricted to the above described cells in the kidney and occurs in many cell types (especially HIF-1α) and HIF binds to other promoters than that on the erythropoietin gene and regulate changes in these cells in response to hypoxia (Schödel and Ratcliffe 2019).
Erythropoietin binds to the erythropoietin receptor on developing erythroblasts, which triggers a signaling cascade mediated through the non-receptor tyrosine kinase (phosphorylates tyrosine residues on other proteins), Janus-activated kinase-2 (JAK2), resulting in the differentiation and survival (by upregulating anti-apoptotic genes, such as BcL-xL) of erythroid progenitors. Mutations in JAK2, primarily at amino acid 617 (in which a valine [V] is exchanged for phenylalanine [F]) cause constitutive activation of the erythropoietin receptor, so signaling occurs in the absence of erythropoietin, driving erythropoiesis in the absence of hypoxia. This causes the neoplastic condition, polycythemia vera or chronic erythrocyte leukemia, a chronic myeloproliferative disorder of erythrocytes. A homozygous JAK2 mutation (V617F) has been identified in up to 90% of human patients with polycythemia vera, indicating it is the most common causative mutation of this hematopoietic neoplasm (Stein et al 2015). A JAK2 mutation has been detected in 1 of 5 dogs with suspected polycythemia vera, indicating conservation of this pathophysiologic mechanism across species (Beurlet et al 2011). Conversely, the absence of erythropoietin results in a RBC production defect, with subsequent anemia. Decreased erythropoietin production in the kidney is thought to be one mechanism responsible for anemia of chronic renal disease (although other factors, such as inflammatory cytokines, can contribute and concurrent blood loss can worsen the anemia). Interestingly, the renal fibroblast-like cells are thought to contribute to fibrosis (through secretion of fibrogenic cytokines) that occurs in (and likely worsens) some renal diseases (Souma et al 2015). Note, that JAK2 is also downstream of the thrombopoietin receptor. Heterozygous mutations in JAK2 (same amino acid – V617F) result in essential thrombocythemia or chronic platelet leukemia (megakaryocytes are more sensitive to JAK2 than erythroid cells).
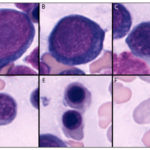
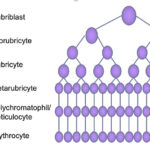
The earliest erythroid progenitor is the blast forming unit-erythroid (BFU-E), followed by the colony forming unit-erythroid (CFU-E). These give rise to recognizable erythroid precursors, proerythroblasts (rubriblasts, prorubricytes), basophilic erythroblast (basophilic rubricyte), polychromatophilic erythroblast (polychromatophilic metarubricyte), orthrochromic erythroblast (metarubricyte), reticulocyte and mature RBC. Note the different terminology is that used in human medicine with terms used in veterinary medicine in parentheses. It is the late CFU-E that have erythropoietin receptors, which are gradually shed as the precursors differentiate, such that they are absent on reticulocytes and mature red blood cells. Unlike general hematopoiesis, erythroid maturation is considered to be tree-like in hierarchy, with one progenitor giving rise to two daughter cells, which are smaller. The most immature cells have the most protein synthetic machinery and have larger nuclei (for DNA synthesis and transcription) and bluer cytoplasm (RNA/ribosomes for translation). As the cells divide, the nucleus becomes smaller and pyknotic and is finally expelled as it is obsolete. Hemoglobin starts being produced and as it accumulates, the need for protein synthesis declines, leading to a decrease in organelles, including ribosomes, in the cell resulting in less blue cytoplasm (that is offset by the color of hemoglobin, so you get a mixture of red and blue, depending on which dominates). The final mature red blood cell just contains large amounts of hemoglobin.
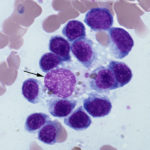
Central to erythropoiesis is the need for cellular coordination of iron acquisition (needed for hemoglobin production) and cell proliferation. Macrophages in the bone marrow provide erythroid precursors with iron, via releasing iron from intracellular stores (ferritin) through a carrier protein, ferroportin. The liberated iron binds to transferrin, which is then taken up by transferrin receptors on the developing erythroid cells via clathrin-mediated endocytosis. The iron-free transferrin is then recycled to the surface of the cell for more iron uptake. Additional scavenger receptors have been identified that also allow erythroblasts to take-up iron that is protein bound. An alternative route for iron uptake is pinocytosis, which has been shown in vitro, but is of unknown relevance in vivo. To acquire the iron, developing erythroid progenitors are clustered around macrophages in the marrow, in so-called “erythroblastic” islands. The interaction between erythroid precursors and macrophages is more than just about iron uptake; macrophages also provide adhesive interactions, such as via erythroblast macrophage protein, CD163 (the hemoglobin-haptoglobin receptor) and VCAM-1 and VLA-4 (on macrophages and erythroblasts, respectively), which facilitate erythropoiesis, and take up extruded nuclei (de Back et al 2014). The process of nuclear extrusion is fascinating, seeming to be a highly co-ordinated event, requiring a process akin to cell division (cytokinesis), differential protein sorting, vesicle movement and autophagy (e.g. of mitochrondria), which results in the formation of a red blood cell fragment containing the nucleus, called a pyrenocyte. During nuclear extrusion, vimentin is downregulated; this intermediate filament is thought to be essential for retention of the nucleus in the cell. Indeed, retention of vimentin may explain the presence of nuclei in red blood cells from birds, reptiles, fish and amphibians. In addition, specific proteins are compartmentalized to the anucleated reticulocyte portion or the nucleated pyrenocyte portion (e.g. VLA-4). Partitioning of VLA-4 to the pyrenocyte makes sense, because this integrin will bind to VCAM-1 on macrophages within the erythroblastic island, facilitating uptake. Uptake of the pyrenocyte by macrophages appears to involve additional mechanisms, including phosphatidylserine exteriorization (with recognition by phosphatidylserine receptors on macrophage) and erythroblast macrophage protein. The phagocytized pyrenocyte and its nucleus are degraded within the macrophage (Keerthivasan et al 2011).
The anucleated erythrocyte that still contains RNA can be detected by supravital dyes that precipitate RNA or dyes that bind RNA. Immature anucleated erythrocytes that have detectable RNA are called reticulocytes. We quantify reticulocytes in dogs and cats to assess the regenerative response by the marrow to an anemia (of hemolytic or hemorrhagic origin). Once a reticulocyte is formed, it stays in the bone marrow for approximately 24 hours (in people, we do not know precisely how long this stage takes in our common domestic species) and then the cell is released into circulation. The plasma membrane composition of the RBC changes to facilitate this release, largely by decreasing the expression of, and cleaving off, adhesion molecules. As reticulocytes mature to erythrocytes within their circulation, they degrade their RNA into nucleotides (which are then broken down to nucleosides by pyrimidine 5′-nucleotidase, which is inhibited by lead, resulting in basophilic stippling or nucleotide retention in erythrocytes), eliminate their mitochondria (potentially through autophagia and exosomes), remodel (losing surface and volume area) to become more flexible, and drop off receptors (such as the transferrin receptor, which is required for iron uptake and hemoglobin synthesis in nucleated erythroid progenitors) (Ney 2011).
Erythropoiesis does not only occur in the steady state or as an expected response to an anemia due to blood loss or red blood cell destruction (hemolytic anemia). Stress erythropoiesis with recruitment of early erythroid progenitors can occur in response to inflammation, tissue hypoxia or failure/suppression of normal steady-state hematopoiesis or erythropoiesis in chronic anemias (“bone marrow failure” syndromes). Stress erythropoiesis appears to primarily occur in the spleen (at least in mice) and is associated with an inflammatory response, including hepcidin-mediated sequestration of iron and cytokine suppression of bone marrow erythropoiesis, with shifting of bone marrow production to myeloid cells (Hattangadi et al 2011, Paulson et al 2020). Stimulation of erythropoiesis under states of stress is facilitated by glucocorticoids and hypoxia-inducible transcription factors (HIF), erythropoietin release from the kidneys, and erythropoietin-mediated release of erythroferrone (which inhibits hepcidin), although the stimulation occurs via different transcriptional signals than that of steady-state erythropoiesis, with macrophages playing a key role. Stress erythropoiesis may occur intermittently in states of inflammation and failure of stress erythropoiesis may contribute to anemia of inflammatory disease. The stimulation of stress erythropoiesis is postulated to promote resolution of inflammation, since erythropoietin may induce anti-inflammatory responses in macrophages (Paulson et al 2020).
Structure
The structure of the RBC is amazingly well-tailored to the complex function these cells have. For the duration of the RBC lifespan, these cells must traverse the blood stream at relatively high speeds and maintain the perfect degree of fluidity to allow them to move through the vessel without breaking and concurrently allow for efficient gas exchange across the cell membrane. This fluidity is accomplished by the complex cytoskeletal structure of RBC, which is enclosed by aphospholipid bilayer, containing numerous transmembrane proteins. The cytoskeleton of the RBC has a high tensile strength, but it also highly deformable. The mechanical properties and shape of the RBC allow the RBC to get through very small capillaries without being damaged, because the diameter of the RBC is larger than the diameter of the smallest capillaries it must traverse. Alterations in the RBC cytoskeleton can lead to RBC fragility (osmotic and mechanical) and abnormal RBC shapes.
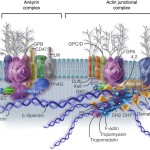
The cell membrane is composed of a lipid bilayer with embedded carbohydrates and proteins, and an underlying cytoskeleton. The membrane lipids are important for maintaining cell shape and surface area. The membrane carbohydrates form blood groups, which are predominantly defined by the types of carbohydrates in the red cell membrane. The membrane proteins form various membrane receptors and transport channels. A number of the proteins are linked, both mechanically and functionally, to the metabolism of the RBC. The location of these proteins and affinity for other proteins can be altered by pH within the cell, oxygenation of hemoglobin, and oxidation. Investigators have postulated that some of the “loosening” on the linkages that occurs with deoxygenation of hemoglobin helps to increase the flexibility of the RBC thus potentially improving blood flow in hypoxic tissues. This could also lead to membrane damage and “aging” of the RBC.
The RBC cytoskeleton consists primarily of the protein spectrin, which forms a sub-membraneous cytoskeletal framework (reviewed by Lux 2016). Spectrin consists of two elongated chains (α and β) which are intertwined and can be stretched or coiled. The spectrin network is associated with other proteins, such as ankyrin and band 4.1. Together, these proteins form a complex lattice-like meshwork that allows the RBC to have incredibly high tensile strength as well as deformability. The spectrin chains are linked to actin and to the RBC membrane via adaptor proteins, such as ankyrin, to band 3, a transmembrane protein that is involved in anion and carbon dioxide exchange.Actin also binds to the membrane bilayer in a complex with protein 4.1, p55 and glycophorins C and D. Defects in the cytoskeleton proteins can lead to a loss of structural integrity of the cell, which can shorten its lifespan or result in visibly-recognizable defects in shape. Defects in spectrin are responsible for the condition of hereditary spherocytosis in human beings. Spectrin deficiency has been identified in asymptomatic Dutch Golden Retrievers , which displayed increased osmotic fragility, but no spherocytes were evident in blood smears (Slappendal et al 2005). A mutant β-spectrin was identified in an asymptomatic dog with elliptocytosis (de Terlizzi et al 2009). A deficiency of protein 4.1 was reported in a dog (Smith et al 1983). As for other reported membrane defects in dogs, RBCs in the dog of this case report were also osmotically fragile. Oxidative injury, which accumulates in RBCs as they circulate over time, alters band 3, rendering it antigenic. Naturally occurring antibodies recognize the altered band 3 and facilitate phagocytosis by mononuclear cells (primarily in the spleen) resulting in the removal of effete RBCs.
Function
The primary functional role of the RBC is to transport oxygen from the lungs to the tissues and carbon dioxide from tissues to the lungs for expulsion. This functional role is served by hemoglobin (Hgb). Each Hgb molecule is composed of 4 iron-containing heme units and 4 polypeptide globin units, consisting of 2 α and 2 β chains. The heme unit is a porphyrin ring and is the portion of the hemoglobin molecule that binds iron. The globin unit consists of polypeptides and functions in part to prevent the iron-containing heme molecules from coming in close contact with each other, which would interfere with function. Oxygen binds reversibly to the iron within the heme molecule and is released in tissues (promoted by multiple factors including 2,3-DPG). Carbon dioxide is transported from the tissues back to the lungs in 3 ways: Some (about 20-30%) is transported reversibly bound to amine groups in hemoglobin (not to iron), a small amount (about 7%) is transported as dissolved CO2 in the blood, and most (about 70%) is transported inside the red blood cells as carbonic acid produced from CO2 and water by carbonic anhydrase. The hemoglobin also binds hydrogen and intracellular Hgb serves as a body buffer or base, protecting against changes in pH.
Hemoglobin also provides viscosity to the RBC cytoplasm. This is crucial in maintenance of cell shape and membrane stability. In states of iron deficiency, the decrease in cytoplasmic hemoglobin is thought to contribute to a decrease in cytoplasmic viscosity, and this is followed by membrane instability (hence we can often see cellular fragmentation, including acanthocytes and keratocytes, in severe iron deficiency).
Because RBCs carry so much oxygen, they are prone to oxidative injury. Normally, as oxygen binds to and is released from the iron in Hgb, superoxide radicals can be formed in small amounts. RBCs have antioxidant mechanisms to deal with this as long as it is not excessive (more on this below). However, exposure to certain exogenous oxidative compounds can overwhelm the RBC antioxidant capability and lead to oxidative damage. Some of the more clinically important oxidative compounds include onions, wilted red maple leaves (horses), acetaminophen, and various other drugs/toxic plants. Endogenous oxidants are produced in disease states include inflammation, diabetes and neoplasia. Lipids in the cell membrane and thiol (SH, sulfhydryl) groups are especially sensitive to oxidation. If membrane lipids/proteins are oxidatively-injured they may stick together and lead to the formation of eccentrocytes. Similarly, if hemoglobin is oxidized and precipitates, this can lead to the formation of Heinz bodies.
Metabolism
Since RBCs lack mitochondria, they can only undergo anaerobic glycolysis (Embden-Meyerhof pathway). The generation of ATP and metabolic intermediates through anaerobic glycolysis allows the RBC to maintain membrane integrity, cell size, and redox status. ATP is required to maintain cell shape and deformability, since it is needed to regulate water and electrolyte content (which affects size and shape of the cell) and to maintain the cytoskeleton (which is needed for proper deformability). As a result, RBCs can lyse when depleted of ATP. The generation of ATP occurs at two main steps of the glycolytic pathway: a) Conversion of 1,3 DPG to 3 PG (this ATP is not produced if the 1,3 DPG is shunted off for 2,3-DPG production, see below) and b) Conversion of phosphoenolpyruvate to pyruvate via pyruvate kinase.
There are three main branch points off the glycolytic pathway that produce various other compounds that are necessary within the RBCs: the hexose monophosphate shut (also called the pentose phosphate pathway), the methemoglobin reductase pathway, and the Luebering-Rapaport pathway (which is commonly referred to as the 2,3- DPG shunt).
- Hexose monophosphate shunt: This helps protect RBCs against oxidative injury. The enzyme, glucose-6-phosphate dehydrogenase (G6PD) produces NADPH from glucose 6-phosphate in the glycolytic pathway, which is utilized to help maintain glutathione (GSH) in a reduced state. Reduced glutathione is an important antioxidant peptide and can protect against oxidation of iron, Hgb sulfhydryl groups, or cell membrane proteins. This pathway is incredibly important in RBC as they lack the organelles other cells use to assist them in dealing with oxidative stress (nucleated cells can alter gene transcription and increase synthesis of proteins that are involved in anti-oxidant and protective pathways, such as the heat shock proteins). Inherited defects in this pathway lead to oxidant injury in RBCs, manifesting as eccentrocytes and Heinz bodies. A presumed congenital defect in G6PD has been reported in a horse (Stockham et al 1994, Harvey 2006 review).
- Methemoglobin reductase pathway: This also helps protect RBCs against oxidative injury, but is focused on hemoglobin oxidation, specifically the iron portion of the heme ring. Methemoglobin (Hgb-Fe3+) is hemoglobin that contains non-functional oxidized ferric iron (Fe3+) rather than the functional form of ferrous iron (Fe2+). Methemoglobin is considered non-functional because it cannot bind oxygen; however, this oxidative change to iron in hemoglobin is reversible. NADH produced in the glycolytic pathway is a cofactor for the enzyme methemoglobin reductase, which reverses the oxidation of iron and returns it to the normal reduced (Fe2+) state. Methemoglobin is now known to consist of two enzymes, cytochrome B5 and cytochrome B5 reductase. Congenital deficiencies in the methemoglobin reductase complex or a cofactor for the enzyme complex, flavine adenine dinucleotide (FAD), have been reported in animals (Harvey et al 2003, Harvey 2006 review. McKenna et al 2014). In one affected mustang mare, supportive laboratory findings were eccentrocytes and methemoglobinemia (manifesting as brown-tinged blood) (Harvey et al 2003). In a dog with a congenital defect in methemoglobin reductase, due to a deletion in the promoter region of the cytochrome B5 gene, the dog presented with severe cyanosis and marked methemoglobinemia (38% methemoglobin, normal <3%) (McKenna et al 2014). Similar clinical signs were seen in an affected kitten with methemoglobin reductase deficiency (unknown genetic defect) (Harvey et al 1994).
- Luebering-Rapaport pathway: This produces 2,3-DPG from an intermediate within the glycolytic pathway. 2,3-DPG is an organic phosphate compound that functions to promote release of oxygen from hemoglobin into tissue. It binds with greater affinity to partially deoxygenated hemoglobin and promotes release of the remaining oxygen molecules bound to the hemoglobin. When 2,3- DPG is bound to Hgb, the dissociation curve of Hgb is shifted to the right. A decrease in pH (acidosis in the tissue, which occurs with increased production of lactate) and an increase in temperature have the same effect on the Hgb saturation curve. Dogs with phosphofructokinase (PFK) deficiency have decreased amounts of 2,3-DPG, because the synthesis of this compound is below the PFK step of the glycolytic pathway (Harvey 2006 review).
Senescence
RBC life spans (the length of time that RBCs circulate in the blood) vary depending on species. The RBC life span in routine domestic species is about 2 (cats) to 4 (dogs) to 6 (horses, cattle) months, but is much shorter is some small mammals (~10 days in gerbils) and much longer in exotics (~600-800 days in eastern box turtles).
In healthy animals, about 1% of RBCs are removed from circulation each day due to normal aging and damage. As the RBCs accumulate damage, they become denser and less deformable, largely as the result of repeated rounds of oxidative stress (remember mature RBC are unable to synthesize any new proteins). This leads to accumulation of oxidant injury to hemoglobin, which promotes accumulation of surface-bound immunoglobulins. These immunoglobulins serve as opsonins and are one of the main signals for splenic macrophages to remove older RBCs from circulation. Aged RBC also invert their lipid membrane, much like nucleated cells undergoing apoptosis, and expose phosphatidylserine on their surface membranes. In RBCs, this process is called eryptosis and it leads to uptake of RBC by macrophages, similarly to uptake of extruded nuclei in pyrenocytes. Another important aging change in RBCs is a decreased ability to produce ATP. This can lead to decreased membrane deformability. If RBCs cannot traverse splenic sinusoids well due to decreased deformability, this signals splenic macrophages to remove them from the circulation. Within the macrophages, the RBCs are degraded. The amino acids from the globin portion of Hgb are recycled and can be used again. The heme portion is broken down into iron and porphyrin, with the latter being converted to unconjugated bilirubin (in those species that have biliverdin reductase) or biliverdin (e.g. birds). The iron is either released to transferrin for transport to marrow or stored as ferritin in the macrophage. Thus, the iron released from degraded effete red blood cells can be used again in the bone marrow for production of new RBCs. Thus, removal of effecte RBCs by macrophages is a form of extravascular hemolysis, but it is physiologic and not pathologic (RBCs are being removed once they have completed their lifespan and not prematurely, since the latter would result in anemia). Normally, a very small amount of intravascular hemolysis (popping of the cells in the blood stream) does occur, when aged or damaged RBCs rupture while still in circulation. This can occur when the RBC are ATP-depleted. The free hemoglobin is then rapidly cleared from circulation by binding to carrier proteins such as haptoglobin. The hemoglobin:haptoglobin complexes are taken up via scavenger receptors, such as CD163, into the macrophages. We would never see such a small amount of liberated hemoglobin as hemoglobinemia, thus any hemoglobinemia occurring in vivo is pathologic, indicating red blood cells are lysing (popping or rupturing) in the circulation due to a red blood cell defect (e.g. phosphofructokinase deficiency), oxidative injury (e.g. red maple leaf poisoning) or the presence of complement or complement fixing antibodies on the RBC membrane (intravascular variants of IMHA).