
Cholestasis can be defined as the stoppage or suppression of bile flow.
There are several laboratory markers of cholestasis. These are:
- “Cholestastic” or “inducible” enzymes alkaline phosphatase (ALP) and γ-glutamyl transferase (GGT). These are bound to cell membranes of biliary epithelial cells and primarily serve as indicators of cholestasis. ALP, however, is less than specific for this purpose, in that its activity can be influenced by a variety of other factors, including induction secondary to corticosteroids in dogs. GGT is more specific in general, and more sensitive in some instances.
- Bilirubin, particularly conjugated bilirubin: When severe enough, cholestasis will result in increased direct bilirubin concentrations in blood, which spills into the urine, resulting in bilirubinuria. ALP and GGT usually have a greater sensitivity for cholestasis compared to serum bilirubin levels alone. For instance, localized cholestasis may increase ALP and GGT concentrations without any increases in serum or urine bilirubin. Cholestasis will result in secondary liver damage (bile acids have an emulsifying effect on cell membranes), leading to increased liver leakage enzymes and an increase in indirect bilirubin as well.
- Bilirubinuria; In all species other than dogs and ferrets, bilirubinuria is abnormal in urine, is due to increased direct bilirubin in blood, and indicates cholestasis. Small amounts of bilirubin (conjugated or direct bilirubin) can be seen in concentrated urine of dogs and in ferrets, but bilirubinuria in excess is still abnormal in the latter species.
- Other abnormalities may be observed in cholestatic animals, but are generally not used as markers of cholestasis (too none specific), e.g. hypercholesterolemia, increased bile acid concentrations (see below under laboratory diagnosis).
Types of cholestasis
The flow of bile is dependent on the active transport of bile acids via protein transporters into canaliculi creating an osmotic gradient and the patency of the biliary tract from the canaliculi to where the common bile duct enters the intestine. Cholestasis can therefore be associated with either a structural obstruction or a “functional” defect (affecting the function of bile acid transporters or osmotic gradient) to bile flow. Cholestasis causes major changes to hepatocytes, including leaky intracellular gaps (through which bile and its constituents can leak or “regurgitate” back into blood) and downregulation or reversal of protein transporters (which also cause regurgitation and decreased uptake of organic compounds, such as bilirubin and bile salts).
Structural cholestasis
Structural cholestasis involve a physical impediment to bile flow. It can be intrahepatic or extrahepatic:
- Intrahepatic obstructions that compresses biliary canaliculi:
- Hepatocellular swelling – e.g. hepatic lipidosis, severe corticosteroid hepatopathy in dogs
- Severe cellular infiltrates (inflammatory or neoplastic), particularly those around the portal area and biliary tree
- Solid tumors – primary to the liver (e.g. cholangiocarcinoma) or metastatic
- Fibrosis around the biliary system – response to chronic inflammation
- Choleliths, parasites (e.g. Fasciola hepatica)
- Bile sludging in canaliculi (e.g. severe dehydration in cats)
- Extrahepatic obstructions affecting the extrahepatic biliary system:
- Tumors – e.g. pancreas, biliary tract, duodenum
- Inflammation, e.g. pancreatitis
- Fibrosis – e.g. secondary to chronic recurrent pancreatitis
- Choleliths
- Gall bladder mucocoeles
“Functional” cholestasis
So-called “functional” cholestasis involve defects in the transporters needed for active transport of bile acids into the canaliculi, although recent evidence indicates that even in structural problems, cholestasis results from downregulated transporters. These are ATP-dependent and excretion is the rate limiting step in bilirubin metabolism e.g. Na/K ATPase. Functional cholestasis can occur if drugs, hormones, cytokines, endotoxins or excess free fatty acids (fatty acid toxicity) interfere with the hepatocyte transporters, responsible for bilirubin excretion (MRP2).
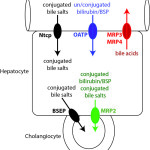
Transporters
Alterations in hepatocyte transporters are thought to be the primary events in mediating hepatocellular cholestasis. Some transporters are present on cholangiocytes, although there is little known of these at present (e.g. uptake of bile salts from bile occurs via an apical or luminal sodium-dependent bile salt transporter; excretion into blood via multidrug resistance-associated protein 3 on cholangiocytes may have a role in returning bile salts to the systemic circulation during cholestasis).
Uptake on sinusoidal or basolateral membrane (from blood)
The following transporters are located on the blood sinusoidal or basolateral aspect of hepatocytes and take up substances from blood into hepatocytes.
- Sodium taurocholate cotransporter (Ntcp): A sodium-dependent-carrier mediated transporter takes up conjugated bile acids (> 80% taurocholate) with sodium and less (<50%) unconjugated bile acids (e.g. cholate). The driving force required for this transporter is provided through basolateral (sinusoidal) Na/K ATPase. Drugs and hormones can influence this Na/K ATPase and decrease bile flow (resulting in cholestasis). Lipopolysaccharide and inflammatory cytokines, such as TNFα and IL-1, can downregulate both Ntcp and Na/K ATPase expression by more than 90 and 50%, respectively, resulting in “functional” cholestasis in sepsis. Bile duct ligation also decreases these transporters.
- Organic anion transporting polypeptide family (OATP): These are responsible for sodium-independent carrier-mediated uptake of unconjugated bile acids, bromsulphthalein (BSP; a dye used in liver function tests), conjugated (and maybe unconjugated) bilirubin and organic anions. Activity of this transporter is downregulated with bile duct ligation and by cytokines (“functional” cholestasis of sepsis). The function of these transporters involves anion exchange with bicarbonate and reduced glutathione (high intracellular concentrations of reduced glutathione stimulates OATP uptake) and some may work in the opposite direction when there are high intracellular concentrations of some proteins (serving to get rid of potentially dangerous compounds, when needed), i.e. responsible for regurgitation of bile acids and “conjugated bilirubin” back into blood in cholestasis.
- Multidrug resistance-associated proteins 3 and 4 (MRP3, MRP4): These are involved in bile acid efflux from the hepatocyte into blood (regurgitation) and are a protective mechanism against the build up of bile acids intracellularly, since they are toxic (emulsify membranes). MRP-3 is normally expressed in low concentrations but is induced in certain conditions, such as inherited defects in canalicular transport proteins (Dubin Johnson syndrome) or biliary cirrhosis in human beings. MRP-4 is involved in secretion of reduced glutathione (GSH) and monoanionic bile salts.
- Other transporters: Unconjugated bilirubin and fatty acids are taken up by, as yet, unknown transporters (the OATP family may be partly involved in unconjugated bilirubin uptake). Fatty acid uptake is dependent on sodium-dependent mechanisms.
There are other transport mechanisms, such as passive ionic diffusion. Some unconjugated bile acids are taken up by this mechanism.
Excretion into the canaliculi (bile)
Bile formation and flow is largely driven by excretion of bile salts, which are osmotically active and create a concentration gradient, which water follows allowing excretion of fluidic bile (called bile salt-dependent flow). Bile acid secretion is linked with excretion of the electrolytes, sodium, chloride, and potassium. There is also bile salt-independent bile flow, which is stimulated by a bicarbonate/chloride exchanger, which actively transports bicarbonate into bile, in exchange for chloride uptake. The latter exchanger is also found in cholangiocytes. Unconjugated bile salts, such as ursodeoxycholate (which is used to treat some cholestatic conditions) promote bile salt-dependent flow through stimulating this transporter.
The active excretion of compounds into the biliary canaliculi is the rate limiting step for bile formation. Excretion is accomplished by the following ATP-dependent transporters, which unlike some sinusoidal transporters, only work one way (that is excretion of solutes into bile and not vice versa).
- Bile salt export pump (BSEP): This sodium-independent transporter excretes bile salts and is the principal force for bile salt-dependent bile flow.
- Multidrug resistance-associated protein 2 (MRP2): This transports conjugated bilirubin and conjugated bile salts into bile and contributes to bile salt-independent bile flow. This is also the transporter that excretes non-bile acid organic acids, e.g. BSP. This transporter is absent in Dubin-Johnson syndrome in human beings and is downregulated by cytokines and bile duct obstruction.
- Multidrug resistance-1 gene (MDR1, P-glycoprotein): This transports foreign substances (drugs), organic cations and cytotoxins into bile.
Defects in these transporters occurs under the following situations:
- Bile duct ligation (extrahepatic cholestasis): Bile salt accumulation injures hepatocytes, so protective mechanisms have developed to decrease bile salt uptake via downregulating Ntcp, the main bile salt uptake transporter on the sinusoidal surface. Unfortunately, one of the bile salt (and bilirubin) transporters on the canalicular membrane is also markedly downregulated, preventing excretion of the accumulating bile salts. The OATP family is also decreased, contributing to hyperbilirubinemia.
- Endotoxins and inflammatory cytokines: These affect sodium-dependent bile salt uptake (Ntcp, sodium/potassium exchanger) and bile salt-dependent (BSEP) and independent (MRP2) bile flow. The bicarbonate/chloride exchanger is also affected, worsening defects in bile salt-independent flow. This results in what is called “functional” cholestasis, which may not stimulate marked increases in the inducible or “cholestatic” enzymes, ALP and GGT (which increase to high levels with extrahepatic cholestasis).
Laboratory diagnosis
The following laboratory findings are helpful for recognizing the presence of cholestasis in an animal:
- Hyperbilirubinemia (conjugated higher than unconjugated)
- Bilirubinuria: Conjugated (direct) bilirubin is water soluble and will rapidly appear in the urine (excessive for USG in the dog, likely ferrets, and abnormal in other species regardless of USG).
- Often disproportionate increases in inductive over leakage enzymes (ALP and GGT will be higher than ALT and AST)
- Note that inflammatory diseases (such as chronic/chronic active hepatitis) can cause cellular injury and concurrent cholestasis due to swelling and or/fibrosis. The enzyme patterns in these disease can be quite heterogeneous.
- Abnormal vitamin K absorption, leading to hemostatic abnormalities: Absorption of fat soluble vitamins is dependent on intestinal emulsification of fats by bile acids. Reduced bile flow results in decreased absorption of these fat soluble vitamins, especially vitamin K. Vitamin K is required for the activation of coagulation factors II, VII, IX and X and the inhibitors, protein C and protein S. This may result in a prolonged PT (may be affected before APTT, because of short half life of FVII) and/or APTT or low activity of FVII:C. May not see clinical bleeding.
- Hypercholesterolemia: This is thought to be due to:
- Decreased clearance of cholesterol in bile
- Increased production of cholesterol-rich lipoproteins:
- Cholestatic diseases are associated with the production of an aberrant lipoprotein (called lipoprotein X). Studies have implicated this lipoprotein as having a role in interfering with the negative regulatory feedback loops that normally reduce cholesterol synthesis when cellular levels are high.
- Decreased lipoprotein clearance
Observations at Cornell University suggest that higher cholesterol concentrations are seen with extrahepatic versus intrahepatic cholestasis.
- Increased bile acid concentrations: Serum bile acids will increase with cholestasis so this test is generally used as an indicator of hepatic function in the absence of cholestasis (and should generally not be run in animals with evidence of cholestasis).