
Abnormalities in platelet function are called thrombopathias (also thrombocytopathy or thrombopathy). They can develop from increased platelet function (resulting in thrombosis) or decreased function (resulting in hemorrhage). Thrombopathias are more commonly recognized in dogs than other species.
Inherited
Inherited thrombopathias should be suspected in a young animal with a bleeding disorder, but showing normal results on coagulation testing (platelet count, PT, APTT, fibrinogen). The buccal mucosal bleeding time as a global test of primary hemostasis may be prolonged in affected patients.
Bernard-Soulier syndrome
This defect is due to a deficiency or abnormality in the GPIb-IX-V complex and results in thrombocytopenia with macroplatelets. Since the complex is required for platelet adhesion to von Willebrand factor under high shear rates (vWf), affected platelets show defective adhesion, but can aggregate normally to usual platelet agonists, such as thrombin (aggregation may be mildly reduced in some human patients) and ADP. However, the snake venom, ristocetin, binds to GP1b-IX-V and triggers platelet aggregation in the presence of vWf. Ristocetin-induced platelet aggregation was used as a functional test for von Willebrand disease, but decreased aggregation in response to ristocetin will also be seen in Bernard Soulier syndrome. The syndrome has been identified in a family of Cocker Spaniels, which demonstrated signs of repeated mucosal bleeding (gingiva, nose) associated with minor trauma (e.g. endoscopy) and post-venipuncture hematomas, manifesting between 2-4 years of age. Bleeding was mild, although severe episodes of hemorrhage did occur after whelping in one dog. The platelet counts in two affected dogs fluctuated between 49-125,000/uL and platelets were large (MPV 24-30 fL, normal usually <15 fL), resulting in a normal platelet crit on some sampling dates. The platelet mean component (granularity) was normal. One dog had a large deletion in the GPIX gene, resulting in a loss of GPIX expression on the platelet surface (Gentilini et al 2019).
Thrombopathia
- Canine thrombopathia: A hereditary, intrinsic platelet disorder that has been described in Basset Hounds and Spitz dogs. Clinical signs consist of chronic mucosal bleeds, petechiae, aural hematomas and prolonged hemorrhage during estrus, shedding of deciduous teeth and after trauma or surgery. The precise mode of inheritance is unknown. The disease is characterized by decreased platelet retention and absent platelet aggregation to all agonists, except thrombin, to which there is a delayed onset and reduced rate of aggregation. Platelet 14C-serotonin release is decreased in response to collagen, but normal to ADP (not in Spitz) and thrombin. Shape change does occur and clot retraction is normal. The defect is now known to involve a platelet signaling molecule called CalDAG-GEF1 (Boudreaux et al 2007), which is a guanine exchange factor that is involved in Rap1 GTP-signaling, which causes inside-out activation of GPIIbIIIa (fibrinogen receptor) in response to agonists (Stefanini and Bergemeier 2014).
- Bovine thrombopathia: An autosomally inherited platelet function defect of Simmental and Simmental-cross cattle, which is also due to a CalDag-GEF1 mutation (Boudreaux et al 2007). Diagnosis is usually made between 2 weeks and 3 years of age. Bleeding episodes are associated with ear tagging, branding or tattooing, dehorning, parturition and surgery. Findings vary from epistaxis, hematuria, hematomas, prolonged estral bleeding and lameness due to subsolar hematomas. Coagulation profiles, platelet counts and platelet concentrations of ADP, ATP and serotonin are normal. There is abnormal platelet aggregation to collagen, ADP, platelet activating factor, calcium ionophores and thrombin.
Glanzmann’s thrombasthenia
Glanzmann’s thrombasthenia (also called thrombasthenic thrombopathia) is an autosomal recessive inherited platelet disorder. This has been recognized in Otterhounds and Great Pyrenees (Boudreaux and Lipscomb 2001) and various horse breeds (Livesey et al 2005, Christopherson et al 2006, Macieri et al 2011, Sanz et al 2011). The defect is due to absent or dysfunctional glycoprotein IIb/IIIa on platelets. GPIIb/IIIa is the platelet fibrinogen receptor and is essential for platelet aggregation (mediated by fibrinogen) and also mediates firm adhesion and spreading on collagen (mostly via binding to vWf, which is bound to collagen). Affected dogs have prolonged bleeding from minor wounds, spontaneous epistaxis and readily form hematomas at sites of injury or venipuncture.
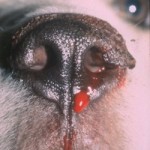
The dogs have a normal to mildly decreased platelet count, normal to increased mean platelet volume and a prolonged BMBT. Decreased platelet retention, absent platelet aggregation to collagen, ADP, platelet activating factor and thrombin, and decreased granule secretion are characteristic. Shape change does occur in response to platelet agonists. Clot retraction is abnormal which helps differentiate this disorder from Bassett Hound thrombopathia. The defect can be in the gene of either glycoprotein IIb or IIIa, as both molecules are required for proper receptor functioning. In both Otterhounds and Great Pyrenees, the defect is due to a genetic mutation that affects a calcium-binding domain of the extracellular portion of glycoprotein IIb (each breed has a different mutation) (Boudreaux and Lipscomb 2001).
Other thrombasthenia
A different disorder has been described in Thoroughbred horses. The expression of the fibrinogen receptor is normal however the receptor binds less fibrinogen and there is a defect in alpha granule secretion. The underlying defect responsible is unknown (Norris et al 2015).
Storage pool diseases
- Chediak-Higashi syndome (CHS): An autosomal recessive genetic disorder characterized by abnormal granule formation in leukocytes, melanocytes, and platelets. Platelets of affected individuals lack discernable dense granules and have deficient or reduced storage pools of adenine nucleotides, serotonin, and divalent cations. Studies of platelet ultrastructure indicate that CHS platelets do not form tight aggregates in response to ADP in vitro. The disease has been identified in a line of Persian cats; all of the affected animals exhibited a “blue smoke” hair color and pale irises with the development of bilateral nuclear cataracts in several individuals. Affected cats experienced prolonged bleeding at incision sites and the development of hematomas following venipuncture. They have prolonged BMBTs and abnormally large granules are observed in peripheral blood granulocytes. Chediak-Higashi syndrome has also been diagnosed in man, mink, mice, cattle, and killer whales.
- Platelet delta-storage pool disease: Reported in American Cocker Spaniels, dogs suffer severe hemorrhage post-venipuncture, surgery or trauma. Coagulation panels, platelet counts and vWf:Ag are all normal. There is a prolonged BMBT, decreased platelet aggregation in response to collagen and ADP and an increased platelet ATP:ADP ratio, due to decreased ADP (mean ATP:ADP of 8.3 compared to a mean ratio of 1.9 in normal dog platelets). Dense bodies are visible on electron microscopy. This disease is attributed to a selective defect in delta (dense) granule ADP transport with deficient ADP storage. Hemorrhage may be severe enough to warrant fresh platelet transfusions.
Acquired
The risk of bleeding in acquired diseases is more unpredictable and typically less severe than inherited disorders. Although abnormal platelet function occurs in these diseases, there may be other causes for the excessive hemorrhage seen in these conditions, such as thrombocytopenia.
Decreased platelet function
Acquired conditions in which there is decreased platelet function include neoplasia, monoclonal gammopathies, infectious disease, hepatic disease, renal disease, pancreatitis, DIC and immune-mediated thrombocytopenia.
- Neoplasia: In essential thrombocythemia and acute megakaryocytic leukemia, platelet aggregation and adhesion are defective. This can also occur in other neoplastic conditions, including chronic myeloid leukemia.
- Monoclonal gammopathy: Very high gamma globulin concentrations associated with some lymphoid or plasma cell neoplasms and atypical responses to some infectious diseases (e.g. Ehrlichiosis) can interfere sufficiently with platelet function in vivo to produce severe hemorrhage. The monoclonal protein (paraprotein) coats platelets, interfering with platelet aggregation, adhesion and phospholipid exposure.
- Infectious agents: Ehrlichia canis and platys can cause decreased platelet aggregation and adhesion (E. platys presumably by activating platelets resulting in platelet exhaustion).
- Hepatic disease: Dogs with various types of hepatic disease have defective whole blood platelet aggregation thought to be due to circulating FDPs, increased bile acids, altered platelet phospholipids and increased proportions of older, less active platelets.
- Renal disease: Mucosal bleeding, reduced platelet retention and a prolonged BMBT are features of natural and experimental uremia in dogs. These abnormalities correlate to the degree of azotemia. Platelet aggregation is either normal or mildly decreased, implicating defective adhesion as the main hemostatic abnormality. The amount and multimeric composition of vWf are normal, indicating that the adhesion defects are not due to vWf abnormalities.
- Immune-mediated thrombocytopenia: Recent studies indicate that some dogs with ITP have abnormal platelet aggregation. This correlated to the immunoglobulin G fraction of the patients’ sera. In human chronic ITP, most antibodies are directed against GPIIb/IIIa and GPIb-IX. Limited canine studies suggest that, in some dogs, there is an antibody against GPIIb/IIIa. As this glycoprotein has an essential role in platelet aggregation, it is not surprising that dogs with ITP will have concurrent platelet dysfunction. In ITP, the clinical signs of hemorrhage do not always correlate to the platelet count, for unknown reasons. It may be that the clinical signs of ITP correlate closer to the degree of platelet dysfunction, rather than the platelet count alone. However, further studies need to be performed in this area.
- Disseminated intravascular coagulation (DIC) and trauma-induced coagulatopathy: In DIC, there is concurrent platelet dysfunction, mediated by FDPs (especially fragments D and E), which compete with fibrinogen for platelet receptors, thus impairing aggregation (Pasqua and Pizzo 1983, Thorsen et al 1986). D-dimer can inhibit platelet aggregation and the release reaction, whereas FDPs (non-crosslinked fragments) can also impair platelet aggregation mediated through glycoprotein VI (Verni et al 2020). D-dimer can bind to activated GPαIIbβ3 and inhibit clot retraction (Buitrago et al 2020). Platelet exhaustion due to excessive activation (such as from DAMPs) is also thought to contribute to thrombopathia in DIC and trauma (Sloos et al 2022).
- Drugs: Drugs that inhibit platelet function include aspirin (which inhibits platelet function by irreversibly acetylating platelet cyclo-oxygenase (COX)-1, thus preventing thromboxane A2 generation, which is needed for secretion and aggregation), older NSAIDs (which reversibly inhibit COX-1 and COX-2 activity) (Luna et al 2007, Mullins et al 2012), combinations of NSAIDs (e.g. dipyrone and meloxicam [Zanuzzo et al 2015]), phenothiazines, heparin and dextran. Trazadone also appears to inhibit platelet function in dogs (Benjamin et al 2023; measured using plateletworks which counts platelets before and after aggregation induced by collagen and ADP) but not horses (as yet unpublished studies from Dr. Colbath at Cornell University; measured with ADP-induced platelet aggregation).
Increased platelet function
Increased platelet function, which may predispose an animal to thrombosis, has been observed in dogs with lymphoma, nephrotic syndrome, and infectious agents (including Rocky mountain spotted fever, heartworm disease and Feline infectious peritonitis). In nephrotic syndrome, the hyperaggregability is thought to be secondary to increased free arachadonic acid availability (for thromboxane production) due to hypoalbuminemia (which normally binds free arachadonic acid).