
Introduction
Definition: Fibrinolysis involves the dissolution of the fibrin clot by the protease, plasmin and takes place in solid phase, i.e. on the surface of cells and fibrin.
Constituents: The fibrin clot, tissue-type plasminogen activator (tPA), contact pathway factors (FXII, prekallikrein, high molecular weight kininogen [HMWK]) and plasminogen. Note that there is also urokinase-type plasminogen activator, but this is mostly seen in tissues and is not thought to be involved in physiologic fibrinolysis to any great extent. It is expressed in an inducible manner by inflammatory cells (e.g. monocytes) and cancer cells and is thought to play a role in pathologic thrombus breakdown (see review on plasminogen by Urano et al 2018).
- Cells: Activated endothelial cells (source of tPA and express bound tPA) and activated platelets (bind plasminogen)
- Enzymes: tPA, plasminogen (proenzyme, plasmin is the enzyme), contact pathway factors (FXII, prekallikrein and high molecular weight kininogen)
- Tissue-type plasminogen activator: This is most efficient at cleaving plasminogen to plasmin when they are bound to insoluble proteins, such as fibrin
- Cofactor: Fibrin, polyphosphates
- Facilitators: Bradykinin (releases tPA)
Fibrinolysis
Sequence of events: Fibrinolysis is also initiated upon vessel injury but is inhibited by thrombin generated through secondary hemostasis. Thus, initially the balance is shifted towards procoagulation, i.e. fibrin production. Once the initiating stimulus for clot formation (vessel injury) is reduced or eliminated (i.e. the defect is sealed by the clot), thrombin generation decreases and the balance then shifts to fibrinolysis.
Sequence of events
This involves the following:
- Release of plasminogen activators.
- Plasmin production from plasminogen
- Clot lysis releasing degradation products.
More information on these events is given below.
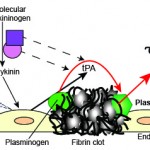
Release of plasminogen activators
Injured endothelial cells express tPA, the main plasminogen activator, on their surfaces and secrete it into plasma. Factor XII is also activated on exposure to subendothelial matrix proteins and forms a complex with high molecular weight kininogen (HMWK) and prekallikrein. This auto-activating complex yields kallikrein from prekallikrein and bradykinin (a potent vasoactive mediator) from HMWK. Bradykinin is a potent inducer of tPA release from endothelial cells and also stimulates release of nitric oxide and prostacyclin from endothelial cells (platelet inhibitors). Furthermore, both FXIIa and kallikrein are direct plasminogen activators but are weaker than tPA.
Plasmin production
Plasminogen binds to fibrin through lysine residues. Plasminogen activators (tPA, FXIIIa and kallikrein) cleave this bound plasminogen to plasmin, which also remains bound to the clot. These activators are inefficient at binding non-bound plasminogen or plasminogen in the fluid phase. Although lysine residues in fibrin are the main binding site, plasminogen can also bind to misfolded proteins and C-terminal residues on cell surface receptors on endothelial cells and activated PS-expressing platelets (GPIIb-IIIa) (Urano et al 2018). The binding of plasminogen to the clot amplifies its conversion to plasmin (fibrin acts like a cofactor for tPA). Plasmin is a potent lytic enzyme, which can cleave proteins other than fibrin. Binding of both plasminogen and its active enzymatic form, plasmin, to fibrin serves to localize fibrinolysis to the clot and prevents plasmin from non-specifically lysing other proteins. The fibrinolytic inhibitor, thrombin-activatable fibrinolysis inhibitor, inhibits fibrinolysis by cleaving the lysine residues from fibrin, resulting in weak plasminogen binding to fibrin.
Clot lysis
Plasmin degrades crosslinked fibrin, releasing variably sized degraded fragments of these proteins, called crosslinked degradation products or X-oligomers. The smallest of these products is D-dimer, which consists of two terminal D-domains of adjacent fibrin monomers joined by γ-γ crosslinks. Plasmin also degrades fibrinogen and soluble fibrin, releasing different degradation products, called fibrin(ogen) degradation products (FDPs). The four main types of FDPs consist of fragments X, Y, E, and D. Lysis of the clot eventually restores vessel patency. All of these degradation products (FDP, D-dimer) are used as laboratory markers of fibrinolysis. Note, that the ability of plasmin to lyse the clot depends on the density and strength of the formed fibrin. Lower concentrations of thrombin forms thinner fibrils and a lens dense network, which is more susceptible to lysis. A thicker or denser network is more resistant to lysis. Polyphosphate, released from dense granules of platelets during activation, help make a dense network of fibrin, that resists fibrinolysis (i.e. is antifibrinolytic).
Regulation
Fibrinolysis is regulated by numerous factors, including inhibitors (see below), the structure of fibrin (thicker strands are more resistant to fibrinolysis [review by Wolberg and Campbell 2008]), clot retraction (resistance to fibrinolysis), and incorporation of RBCs into the clot (resistance to fibrinolysis). Activated protein C is also profibrinolytic (inhibits plasminogen activator inhibitor-1) (Longstaff and Kolev 2015 review).
Inhibitors
Since thrombosis is so infrequently identified clinically, fibrinolytic inhibitors are rarely used therapeutically.
- Physiologic:
- Thrombin-activatable fibrinolysis inhibitor (TAFI, also known as carboxypeptidase B): This is produced in the liver. It is activated by thrombin binding to thrombomodulin on endothelial cell surfaces (the thrombin burst). It is also found in platelets (released with activation – platelets also express thrombomodulin) (Urano et al 2018). This enzyme cleaves lysine residues in fibrin preventing plasminogen binding.
- Plasminogen activator inhibitor-1 and -2 (PAI-1, PAI-2): PAI-1 is the main inhibitor of tPA and is produced in the liver. It is also stored in α-granules in human platelets. It is actually produced in excess of plasmin and binds to endothelial cell-expressed tPA but is limited in its ability to inhibit tPA bound to fibrin (Urano et al 2018). Human patients lacking PAI-1 bleed uncontrollably as they cannot inhibit fibrinolysis (clots break down rapidly).
- Antiplasmin: This is the main inhibitor of plasmin and forms complexes with plasmin, targeting it for degradation. It is less effective when plasmin is bound to the fibrin clot. Factor XIII actually crosslinks this inhibitor to the fibrin, where it may be actively involved in inhibiting fibrinolysis and preventing early clot breakdown (Urano et al 2018).
- Polyphosphates: These are released from dense granules in platelets during the release reaction and make a more rigid dense fibrin clot, that resists fibrinolysis.
- Pathologic: Pathologic inhibitors have not been recognized in animals, likely due to a lack of diagnostic assays for evaluation of this aspect of hemostasis. However, there are many newly recognized inhibitors of fibrinolysis, which are likely more operative in disease states. This includes polyphosphates (see above – these can be released by cancer cells and are also found in bacteria), extracellular nuclear material (released by neutrophils in a process called NETosis, dying cells, and cancer cells) and other neutrophil constituents.
- DNA/histones: These act as competitive inhibitors with plasminogen for binding to fibrin (Urano et al 2018). Cell free DNA also binds to fibrin degradation produces and is a component of the fibrin clot which makes it more resistant to fibrinolysis. The DNA can also help PAI-1 inhibit the action of tPA on generating plasmin from plasminogen. Histones and DNA also make thicker fibrin strands, which are more resistant to fibrinolysis (Gould et al 2015).
- Neutrophil contents: α-defensins released by activated neutrophils are antifibrinolytic (Abu-Fanne et al 2019).
- Pharmacologic: Tranexamic acid and ε-aminocaproic acid. These both act like lysine and are competitive inhibitors for plasminogen (which binds to lysine residues in the fibrin clot). The anti-fibrinolytic activity of these drugs have been evaluated in horses using tPA-induced fibrinolysis with a TEG® (Fletcher et al 2013) and dogs (Fletcher et al 2014, Brown et al 2016).
Clinical signs
Disorders of fibrinolysis are infrequently recognized, mostly due to the lack of sensitive and specific diagnostic assays for this process of hemostasis. Excessive fibrinolysis will result in hemorrhage from clots being lysed too quickly, whereas inhibition of fibrinolysis will cause persistence of a clot, i.e. thrombosis.
- Inadequate fibrinolysis: This is usually due to excess inhibition by PAI-1, rather than decreased concentrations or dysfunction of tPA or plasmin. This is most frequently seen in human patients with sepsis-induced disseminated intravascular coagulation (DIC) and results in thrombosis.
- Excessive fibrinolysis: This is usually a consequence of disseminated intravascular coagulation (DIC) and is attributed to excess plasmin generation, which rapidly destroys fibrin clots leading to hemorrhage. Dogs have far more active fibrinolytic enzymes than humans (Cade et al 1975, Lang et al 1993, Fletcher et al 2014), which may explain why DIC usually manifests as bleeding versus thrombosis.
Sample collection
Several different tubes may be used to measure different aspects of fibrinolysis:
- EDTA-anticoagulated plasma: D-dimer.
- Citrate-anticoagulated plasma: Plasma FDP, D-dimer, all other assays.
- FDP collection tube: Serum FDP. This specialized collection tube contains a snake venom, Botrox atrox venom (Reptilase), to cause clotting and inhibitors of fibrinolysis (aprotonin or soyabean trypsin inhibitor).
Please refer to the sample collection guidelines on how to collect samples appropriately to optimize coagulation test results.
Tests
- Screening tests: FDP assays, D-dimer.
- Specific tests: Plasminogen concentration, tPA activity.
- Tests for inhibitors: PAI-1 activity (Wong et al 2017), tPA-triggered thromboelastography (Fletcher et al 2013, Fletcher et al 2014, Spodsberg et al 2013), TAFI activity (Jessen et al 2010)
- Specialized tests: Plasmin-antiplasmin complexes, euglobin lysis time.
Disorders
Due to the lack of specific diagnostic tests, fibrinolytic disorders are not frequently recognized in animals. However, a syndrome of excessive bleeding in Greyhounds (e.g. severe post-surgical bleeding) responds to tranexamic acid, suggesting increased fibrinolysis is causing the hemorrhage (these dogs have normal coagulation test results, platelet function and platelet count) (Birbeck et al 2019). Other acquired disorders, such as trauma, parvovirus infection, and Angiostrongylus vasorum infection have been associated with enhanced fibrinolysis, however the main disorder is DIC, as indicated above. Reduced fibrinolysis has been detected in dogs with disorders associated with thrombosis (Spodsberg et al 2103). Inherited disorders of fibrinolysis have not been reported and there are only isolated reports of acquired fibrinolytic defects, e.g. parvovirus infection.