
Interpretation
Hepatic copper accumulation with cholestasis and inflammation with secondary Fanconi-like syndrome.
Explanation
Examination of the modified Wright’s-stained smears revealed several clusters of hepatocytes with mild to moderate amounts of small, refractile, turquoise to sea-green cytoplasmic granules, compatible with copper (Figure 1). There were also increased numbers of neutrophils with fewer macrophages, supporting concurrent inflammation. Macrophages contained lipofuscin and hemosiderin in their cytoplasm. Rare necrotic hepatocytes and a few bile casts were observed within some hepatocyte clusters (not shown). Some hepatocyte clusters were affiliated with aggregates of smudged, difficult to discern cells (fibrosis, presumptive), with interspersed bile globules and inflammatory cells. The cytologic diagnosis was copper accumulation with cholestasis, individual cell necrosis, and inflammation. Since the copper was readily identified within hepatocytes and there was evidence of inflammation and necrosis, the copper was considered to be the likely cause of the liver injury (increased transaminase activities, particularly ALT) and cholestasis (increased direct bilirubin with bilirubinuria and bile casts, increased ALP activity). The increased indirect bilirubin could be due to hepatic injury (Questions 1 and 2). An alternative explanation for the high ALP activity is endogenous corticosteroids (stress), since the GGT activity is within reference intervals. Considering the evidence of azotemia (increased creatinine concentration with a 1.020 USG) and tubular injury (proteinuria excessive for the USG, glucosuria without hyperglycemia, granular casts), the high normal urea nitrogen concentration suggested concurrent hepatic dysfunction, with decreased urea synthesis.
|
|
|
|
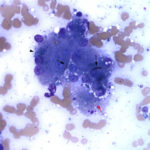
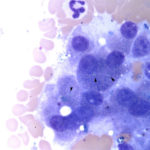
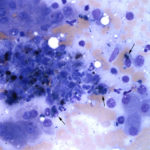
The urinalysis results, specifically the excess proteinuria and glucosuria without hyperglycemia, indicate proximal renal tubular dysfunction (filtered proteins and glucose are primarily resorbed in the proximal convoluted tubules) or acquired Fanconi-like syndrome. In this case, the proximal tubule dysfunction was likely due to copper accumulation in the tubules (this was not confirmed by histopathologic evaluation of the kidneys) (Question 3).
Additional Diagnostic Testing
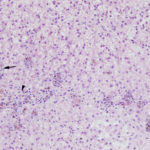
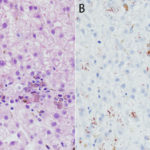
A coagulation panel revealed a mildly prolonged prothrombin time (16.3 seconds, reference interval 11.0-15.5 seconds) and normal activated partial thromboplastin time, further suggestive of impaired hepatic synthetic function resulting in low factor VII activity. Serological testing for leptospirosis was negative and a urine culture was negative. A laparoscopic liver biopsy was performed and submitted for histological evaluation. Assessment of H&E-stained smears of formalin-fixed paraffin-embedded sections of the medial liver lobe revealed multiple copper granulomas (copper within macrophages) scattered throughout the hepatic lobules (Figure 3) with the majority surrounding central veins (zone 3). Lymphoplasmacytic portal infiltrates and mild suppurative infiltrates were noted associated with copper-laden hepatocytes, which were found randomly throughout the lobules, but most were in the centrilobular zone. The presence of copper was confirmed with rhodanine staining (Figure 4) and portal and mild dissecting fibrosis was identified on a reticulin stain. Most of the hepatocytes displayed cytoplasmic distention, compatible with glycogen accumulation. The histopathologic diagnosis was a mild lymphoplasmacytic to suppurative hepatitis with pathologic copper accumulation and a mild diffuse glycogen-type vacuolar hepatopathy (a non-specific finding that may be due to chronic endogenous corticosteroids). Qualitative histological grading for copper using digital image scanning and image analysis software showed a grade 3 out of 5 copper accumulation, i.e. between 50-75% of hepatocytes within all lobules contained moderate to large amounts of copper granules primarily within zone 3. Copper quantification performed in the Toxicology laboratory in the Animal Health Diagnostic Center revealed pathologic amounts of copper (2,118 ug/g, reference interval, <400 ug/g on a dry weight basis).
|
|
|
Discussion
Copper is essential for various biological processes such as mitochondrial respiration, antioxidant defenses, neurotransmitter synthesis, and iron metabolism.1 As a transition metal, copper can cause oxidative tissue damage via Fenton and Haber reactions.2 Dietary copper is absorbed in the gastrointestinal tract (stomach and small intestine) via copper transporters (CTR1) on the apical border of enterocytes.1 Copper then exits enterocytes, via an ATP7A transporter on the basolateral surface, and enters the portal circulation. Within the liver, copper is either stored or excreted in bile with the remainder being distributed via the circulation to other tissues.1,2 Due to its tendency to form superoxide hydroxyl radicals, intracellular copper is never free within and is bound by intracellular scavenger proteins like metallothioneine (MT).2 Within hepatocytes, copper chaperones are responsible for transporting copper for incorporation into proteins, such as superoxide dismutase and glutathione.1 There are two important copper transporter ATPases, known as ATP7A (present in various cell types) and ATP7B (more specific to hepatocytes), that are involved in mobilizing copper to peripheral tissues and biliary excretion.1 Copper is distributed to peripheral tissues bound to the copper-binding protein, ceruloplasmin, and to a lesser extent albumin.1 At low copper concentrations, the transporters with bound copper are localized in the trans-Golgi network. At high copper concentrations, the transporters translocate copper to lysosomal endosomes for biliary excretion.1,2
An overabundance of copper within the liver can cause hepatocellular injury via oxidative damage to cellular and mitochondrial membranes. This injury results in hepatic necrosis, inflammation, fibrosis, and eventually cirrhosis.3 There are 3 mechanisms by which excess copper can accumulate within the liver:
- Defective copper transporters: This is classified as a primary copper storage disease and the copper accumulates due to lack of biliary excretion. This is called Wilson disease in human beings.2 A genetic mutation causing a syndrome similar to Wilson disease has been confirmed in Bedlington Terriers.2,4 Affected dogs have a deletion in exon 2 of the COMMD1 gene, which is responsible for returning the ATP7B transporter back to the trans-Golgi network after delivering copper to the lysosomal endosome for biliary excretion. The lack of biliary excretion results in copper accumulation within hepatocytes.1 Labrador Retrievers, West Highland White Terriers, Doberman Pinschers, Dalmatians, and Sky Terriers are considered to have a predisposition for copper hepatopathy.3 however genetic defects have not been conclusively identified in these breeds. A genome-wide association study in 235 Labrador Retrievers identified missense mutations in ATP7A and ATP7B genes that appear to be linked to pathologic copper accumulation.5 In the latter study, copper uptake and retention assays with canine dermal fibroblasts collected from dogs with a missense mutation (threonine to isoleucine at amino acid 327) in ATP7A showed higher copper levels in cells with the mutation (T327I) than in cells lacking the mutation. In addition, an ATP7B variant (arginine to glutamine at amino acid 1453) remained localized to the endoplasmic reticulum and was not translocated to lysosomes for export under high copper conditions, when transfected into HeLa cells.5 These results suggest copper transport defects may contribute to copper accumulation in Labrador Retrievers.
- Excess dietary copper: This could be a contributing factor to the increased prevalence of copper-associated hepatitis in Labrador Retrievers. Increased amounts of hepatic copper were found in Labrador retrievers with and without chronic hepatitis when biopsied between 1998 to 2010 versus a cohort biopsied between 1980 to 1997.6 The increase in hepatic copper concentrations appeared to coincide with the transition of commercial pet foods from cupric oxide with low bioavailability (<5%) to more bioavailable copper, such as copper sulfate (60% bioavailability).6 The absolute dietary requirement for copper in Labrador Retrievers is not currently known, but based on requirements established for Bedlington Terriers, the ideal copper levels are likely around 5 mg/kg on a dry weight basis.4 The Association of American Food Control Officials (AAFCO) recommend dietary copper levels of approximately 7.3 mg/kg on a dry weight basis in all dogs, which may be high for predisposed breeds.6 Addition of vitamin and mineral premixes may also serve to increase the dietary content of copper.6
- Cholestasis: Unlike cats and human beings, dogs appear to be resistant to this secondary form of copper accumulation.4
Different stages of disease have been determined for Bedlington Terriers, the first dog breed in which a copper hepatopathy was recognized as a primary condition. Affected animals could present with peracute disease and these patients are clinically unstable, with death often ensuing rapidly. Peracute disease is due to acute hepatic necrosis, resulting in copper release into the circulation and an oxidant-induced hemolytic anemia. Dogs surviving the initial insult may remain asymptomatic for years or relapse with less severe signs. Dogs with chronic disease were middle aged or older and had slowly progressive hepatic disease that may acutely decompensate in the face of stress. Clinical signs ranged from vomiting and anorexia to ascites (high or low protein transudate secondary to cirrhosis). The third stage consisted of asymptomatic dogs where the only abnormalities were increased liver enzyme activities, thus the diagnosis must be made by liver biopsy.4
Detection of copper granules within hepatocytes on cytological assessment of liver aspirates is suggestive of copper accumulation. However, the granules can be difficult to see, particularly when in small amounts, and must be distinguished from lipofuscin. Copper tends to be more crystalline (refractile), turquoise versus blue-green and slightly out of the plane of focus compared to lipofuscin. Rhodanine staining can be performed on unstained or previously Wright’s-stained slides to confirm copper accumulation in hepatocytes. In one study of 40 retrospective canine cases, copper was missed on initial cytologic assessment and was only detected on rhodanine staining.7 In the latter study, the authors established a cytologic grading scheme for copper in rhodanine-stained cytologic smears and compared it to quantitative assessment of copper on a dry weight basis in 67 prospectively collected cases. They found that their scheme was relatively sensitive (75%) and specific (97%) for excess copper at dry weight concentrations > 600 ppm, with higher sensitivity (100%) and similar specificity (97%) at concentrations >1,500 ppm. However, the presence of copper within hepatocytes does not always indicate a copper hepatopathy or that the copper is causing liver injury. Furthermore, due to sampling variation, a region with lower amounts of copper may be aspirated, yielding a false negative diagnosis. It is also difficult to confirm hepatic inflammation secondary to injury from copper accumulation from a liver aspirate. This is because most hepatic aspirates are blood-contaminated and it is difficult to detect low numbers of inflammatory cells in excess of that present due to blood. Low numbers of hepatocytes or poor quality smears will also preclude cytologic evaluation of copper stores. Cytologic assessment also does not provide information about zonal localization and hepatic architecture. Moderate or large amounts of copper detected on hepatic aspiration would support the need for more detailed copper assessment via a biopsy, however a biopsy should be pursued if a copper hepatopathy is suspected and copper is not detected on an aspirate (due to sampling variation issues). Note that in this case, carprofen administration was a confounding issue. Carprofen can cause hepatotoxicity,8 however copper accumulation is not associated with carprofen toxicity, which is characterized by vacuolar change, ballooning degeneration, hepatocellular necrosis and mixed inflammation.8 Necrosis in this case was more prominent on cytologic than histologic assessment of the liver.
A liver biopsy is the only way to definitively diagnose copper hepatopathy. Wedge biopsies are preferred over needle core biopsies because they provide a more representative sample and have less architectural artifact associated with sampling.4 Histopathological evaluation provides information about zonal localization, severity of copper accumulation, and distribution and severity of inflammatory lesions.3.4 Copper distribution relative to inflammatory foci can be useful for distinguishing between primary and secondary causes of copper hepatopathy. Primary disease is considered if copper is observed in hepatocytes and macrophages within and adjacent to inflammatory lesions, within hepatocytes distant to inflammatory foci, and within regenerative nodules. Primary copper hepatopathy is also associated with higher amounts of copper in the liver (>2,000 ug/g versus ~1,000 ug/g on a dry weight basis for a secondary hepatopathy). Secondary copper accumulation is generally associated with a periportal distribution.4 Copper concentrations are not always associated with the presence of inflammation. For instance, Doberman Pinschers can have hepatitis with copper concentrations around 600 ug/g on a dry weight basis while Bedlington Terriers may not have any evidence of inflammation with higher copper levels (> 1,500ug/g on a dry weight basis).4 The presence of copper in biopsies should be qualitatively graded with rhodanine staining because this guides therapy and helps with determination of primary or secondary disease. Copper scores range from 0 (no detection of copper granules) to 5 (panlobular copper distribution). For example, histological grades between 3 (as in this case) and 5 warrant therapy, while treatment may not be recommended for grades below 3.2,4 The gold standard for quantifying copper concentration is atomic spectroscopy, although digital scanning of rhodanine-stained smears of a liver biopsy, as done in this case, is gaining preference.9 This method utilizes a positive-pixel algorithm to calculate copper concentration and shows an excellent correlation (R2=0.97) to atomic spectroscopy.9 Digital scanning is advantageous because copper is quantified from the same specimen used for histological grading.
Dogs with copper hepatopathy are treated with copper chelators, like D-penicillamine. Adjunctive hepatoprotectorants are given, including anti-oxidant, anti-inflammatory and antifibrotic drugs. The duration of chelation therapy depends on the severity of copper accumulation. Dogs with hepatic copper concentrations ≥600 ug/g on a dry weight basis are generally treated for 2-6 months or longer in some cases.4 Therapeutic efficacy is best assessed with sequential liver biopsies,2,4 but this is impractical and costly. Sequential monitoring of liver enzyme, particularly ALT, activity is a good method for assessing efficacy.2,4 For chronic therapy, chelation is frequently reduced by 50% or given in alternate day full doses.4 Lifelong dietary copper restriction with subsequent chelation therapy is essential for limiting copper overload and promoting copper removal from the livers of dogs with copper hepatopathy.2,4
Further information and follow up
The dog was initially treated with intravenous fluids, antibiotics, a liver protectant (N-acetylcysteine), vitamin K, and antinausea drugs. After diagnosis, the dog was treated with the copper chelator, D-penicillamine, vitamin B6 (deficiency is documented in humans taking D-penicillamine and may occur in dogs10), antioxidants (vitamin E and S-adenosylmethionine [SAMe] to counteract the oxidative effects of copper), ursodeoxycholic acid (to protect against accumulated bile acid toxicity10), phosphatidylcholine (antifibrotic, antioxidant10), glucocorticoids (to reduce lymphoplasmacytic inflammation11), and a copper-restricted diet. Repeat blood testing 6 weeks later showed complete resolution of the biochemical and urinalysis abnormalities.
References
- Xiaoyan W., Leegwater P., Fieten H. Canine Models for Copper Homeostasis Disorders. Int. J. Mol. Sci. 2016;17:196.
- Dirksen K., Fieten H. Canine Copper-Associated Hepatitis. Vet Clin Small Anim 2017;47:631-644.
- Smedley R., Mullaney T., Rumbeiha W. Copper-Associated Hepatitis in Labrador Retrievers. Vet Pathol 2009;46:484-490.
- Center S. Copper-Associated Hepatopathy. The Merck Veterinary Manual. 10th edition, Whitehouse Station, NJ:Merck & Co;2010:424-428.
- Fieten H., Gill Y., Martin A. et al. The Menkes and Wilson disease genes counteract in copper toxicosis in Labrador retrievers: a new canine model for copper-metabolism disorders. Dis Model Mech. 2016;9:25-38.
- Johnston A., Center S., McDonough S. Hepatic copper concentrations in Labrador Retrievers with and without chronic hepatitis:72 cases (1980-2010). JAVMA 2013;242:372-378.
- Moore R., Coffey E., Hamar D. Diagnostic accuracy of Wright-Giemsa and rhodanine stain protocols for detection and semi-quantitative grading of copper in canine liver aspirates. Vet Clin Pathol 2016;45:689-697.
- MacPhail C., Lappin M., Meyer D., Smith S., Webster C., Armstrong P. Hepatocellular toxicosis associated with administration of carprofen in 21 dogs. JAVMA 1998;212:1895-1901.
- Center S., McDonough S., Bogdanovic L. Digital image analysis of rhodanine-stained liver biopsy specimens for calculation of hepatic copper concentration in dogs. AJVR 2013;74:1474-148.
- Bonagura J., Twedt D. Copper Chelator Therapy. Kirk’s Current Veterinary Therapy XV. Columbus,OH:Elsevier;2013:1624-1625.
- Rifkin J., Miller M. Copper-associated hepatitis in a Pembroke Welsh corgi. Can Vet J 2014;55:573-576.
Authored by: Jorge Adarraga (class of 2017) and T Stokol.