
Interpretation
Oxidative hemolytic anemia, presumptive copper toxicosis
Explanation
Blood smear examination revealed moderate anisocytosis, mild polychromasia, few macrocytes, moderate basophilic stippling, and few Heinz bodies. In addition, there were low numbers of ghost red blood cells (ghost cells) with prominent Heinz bodies (Question 1, Figure 2a). Heinz bodies indicate an oxidative hemolytic anemia, with a component of intravascular hemolysis as seen by the ghost cells and red-brown urine (hemoglobinuria, presumptive) (Question 2). Differential diagnoses for Heinz body formation in goats include copper toxicosis, consumption of oxidant-containing plants, such as onions, garlic, or Brassica species, and selenium deficiency.1 Based on the lack of access to oxidant-containing plants, the results were suspicious for copper toxicosis (Question 3). Exposure of erythrocytes to oxidant toxins, medications, or chemicals can lead to oxidation of sulfhydryl groups on hemoglobin causing the formation of disulfide bonds. The hemoglobin becomes denatured and precipitates to form Heinz bodies. Red blood cells containing Heinz bodies are prematurely removed from the circulation by macrophages, presumably in the spleen (extravascular hemolysis). Oxidative damage to red blood cell membranes also occurs, resulting in increased red blood cell fragility with subsequent intravascular hemolysis.2 Oxidative injury to the iron within hemoglobin (Fe2+ to Fe3+) will form methemoglobin (methemoglobinemia), which would explain the brown color of the doe’s mucous membranes.
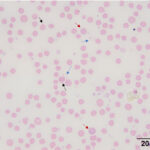
Hematologic findings revealed a macrocytic, hyperchromic, regenerative anemia. The macrocytosis can be attributed to a regenerative response, as seen by the polychromatophilic and macrocytic red blood cells in the blood smear (both of these findings would contribute to the anisocytosis). Basophilic stippling can also be attributed to the regenerative response. Nucleated red blood cells can be seen in regenerative anemia, although the mechanism is unclear (altered bone marrow environment, concurrent bone marrow injury?). Hemoglobin synthesis stops in erythrocyte precursors when an optimal hemoglobin concentration is reached within the cytoplasm.3 Therefore, it is not possible to produce hyperchromic red blood cells. The most likely causes for the mild artifactual increase in MCHC are the intravascular hemolysis and Heinz bodies (Question 4). Intravascular hemolysis would result in a hemoglobin concentration that is higher than the hematocrit due to lysis of red blood cells (MCHC = Hemoglobin ÷ hematocrit). Large Heinz bodies falsely increase the hemoglobin concentration (because Heinz bodies scatter light, which is how hemoglobin is measured). There was also evidence of inflammation or stress (glucocorticoid response), as seen by the leukocytosis which was due to a mature neutrophilia.
Biochemical findings indicated acute liver injury as shown by increased activities of leakage enzymes (AST, SDH, GLDH – although we lack reference intervals for the latter, the GLDH activity is increased compared to goats without liver injury). Note the CK activity was normal in this goat, indicating a liver source for the AST activity. In addition, the hemolytic index from our biochemical analyzer was 64 units, equivalent to mild hemolysis. Release of AST from lysed red blood cells was unlikely to be contributing to the high AST activity. The high GGT activity indicated cholestasis and/or biliary hyperplasia. Cholestasis, which may be secondary to copper-induced damage to the liver, was supported by the mild increase in direct bilirubin. The icteric plasma was mainly due to increases in indirect bilirubin, which was due to the hemolytic anemia. The fact that the hemolytic index was only mildly increased and hemolysis was not visualized in the supernatant plasma from the hemogram indicates that most of the free hemoglobin from lysed red blood cells was being cleared by the kidney in vivo, illustrated in the brown-red urine. The high creatinine concentration could be secondary to a prerenal azotemia from the dehydration, however acute kidney injury was suspected due to the intravascular hemolysis (hemoglobinuric nephrosis). The normal urea nitrogen concentration can be explained by excretion of urea into the gastrointestinal tract, where it is degraded by amino acids by gastrointestinal bacteria flora.
Additional tests
An abdominal ultrasound was performed. The liver appeared markedly heterogenous and mottled bilaterally. In addition, bilateral pyelectasia was evident. A serum copper concentration was mildly increased at 1.25 ppm (reference interval, 0.7-1.2 ppm).
Follow-up
Despite supportive treatment, the anemia persisted and clinical signs worsened. Biochemical changes showed a worsening azotemia (increased urea nitrogen at 44 mg/dL, creatinine, 1.5 mg/dL) and more severe liver injury (AST, 3815 U/L, SDH, 111 U/L, GLDH, 1160 U/L) and cholestasis (direct bilirubin, 1.6 mg/dL). Unfortunately, due to worsening clinical signs, the goat was humanely euthanized.
A necropsy examination was performed, which revealed icterus in the pinnae, mucous membranes, subcutaneous tissue, adipose tissue, and omental fat. The liver was diffusely pale, yellow green and friable. Both kidneys were diffusely dark grey-brown. The urinary bladder contained 32 mL of clear, dark red-brown urine (pigmenturia). Furthermore, the abomasum contained approximately 60 mL of fluid with 0.1-0.2 cm long, dark grey, cylindrical metal fragments, consistent with copper oxide wire particles. Histopathologic examination revealed severe diffuse hepatic necrosis with intracytoplasmic copper within hepatocytes, mixed inflammatory infiltrates (macrophages, neutrophils, lymphocytes), bridging fibrosis (indicating chronicity), bile plugs (cholestasis) and biliary hyperplasia, corroborating the interpretation of the liver biochemical changes. There was attempted regeneration (karyomegalic nuclei). Copper accumulation was confirmed with a Rhodanine stain, which showed that approximately 50% of viable hepatocytes contained small amounts of copper, while dying hepatocytes contained large numbers of copper granules. Cortical and medullary tubules in the kidney were dilated and torturous and numerous tubules contained protein casts with interwoven leukocytes and sloughed epithelial cells (tubular inflammation and tubular injury forming cellular casts) and hemoglobin casts (confirmed by an Okajima’s stain for hemoglobin). In focal areas, there was mineralization of the tubules (presumptive tubular necrosis). Red-brown granules were seen in the renal tubular cells (combination of copper and hemoglobin) (hemoglobinuric nephrosis). The gross and histopathologic findings were consistent with copper toxicosis.
Discussion
Copper is integral in melanin production, elastin and collagen synthesis, iron metabolism, and neurotransmission, making it an essential nutrient for animals.4 Although a necessary nutrient, it can be highly toxic in excessive amounts. Copper toxicity is common in sheep and young cattle but does not occur as frequently in goats.1
There are several recognized forms of the disease, including primary chronic and acute copper toxicity and secondary phytogenous and hepatogenous copper toxicity.1 Chronic copper toxicosis occurs when large hepatic stores of copper accumulate over time as a result of defects in hepatic metabolism, altered biliary and urinary excretion of copper, and/or nutritional imbalances with other trace elements (molybdenum and sulfate).5 In this chronic form, hepatic accumulation of copper causes hepatocellular damage during what is referred to as a prehemolytic stage. This stage is usually subclinical. The hemolytic phase of copper poisoning occurs when large amounts of copper are suddenly released from hepatic stores. This is usually precipitated by a stressful event such as pregnancy, transportation, handling, and/or disease,5 which can trigger the breakdown of copper-containing lysosomes. The result is severe hepatocellular injury and release of copper from the liver into the circulation. The sudden increase in blood copper concentrations causes oxidative damage to red blood cells, inducing Heinz body and methemoglobin formation. Lipid peroxidation of red blood cell membranes and intravascular hemolysis of damaged red blood cells also occurs, resulting in a severe acute anemia. The massive release of hemoglobin can result in hemoglobinuric nephrosis and acute renal failure. Clinical signs of chronic copper toxicity include lethargy, anorexia, rumen stasis, recumbency, icterus, and hemoglobinuria, several of which were seen in this doe.5
Acute copper toxicity, which is less common than chronic toxicity, usually occurs after ingestion of excessive amounts of copper, such as in anthelmintics, disinfectant footbaths, fertilizers, mineral supplements, or poorly formulated feeds.5 Rapid onset of severe gastroenteritis due to necrosis of gastrointestinal mucosa is associated with the acute form of this disease.4,5 Other clinical signs of acute toxicity include emesis, anorexia, dehydration and shock. Intravascular hemolytic anemia and hemoglobinuria may develop after a few days if the animal survives the initial gastrointestinal event.5
Unlike primary copper toxicosis, secondary copper toxicity does not result from an excessive intake of copper. Phytogenous copper toxicity is observed after an animal ingests certain plants (e.g. Trifolium subterraneum) which create a mineral imbalance causing excessive copper retention.6 Hepatogenous copper toxicosis results when an animal ingests plants which cause liver damage and, secondarily, increased retention of copper. Examples of these plants include Heliotropium europaeum and Senecio species, which contain hepatotoxic alkaloids.7
In acute copper toxicosis, a diagnosis can be made by the presence of increased fecal (8,000–10,000 ppm) and renal (>15 ppm by wet weight) copper levels. In cases of chronic copper toxicosis, copper concentrations in serum and liver can be increased.6 A diagnosis can be made if only serum copper concentrations are measured and are increased.8 In this doe, the serum copper concentration was only mildly increased, however the result still supported the diagnosis, based on the clinical signs. It is likely that liver concentrations, if measured in the doe, would have been markedly increased, based on the histologic findings. Serum copper concentrations are not as reliable as liver concentrations for the diagnosis of copper toxicity,8 but it is difficult to obtain liver copper concentrations antemortem (requiring a decent-sized liver biopsy). Necropsy findings associated with copper toxicosis include gastroenteritis with erosions and ulcerations in the abomasum. Tissues may be discolored due to icterus and methemoglobin. Characteristics of this disease include gunmetal blue kidneys with port wine colored urine and a liver that is enlarged and friable. Centrilobular hepatic and renal tubular necrosis are observed upon histological examination.6 Many of these features were seen in this doe.
In conclusion, this case is an example of a rare condition, primary acute copper toxicity in a young goat. This was confirmed on necropsy by the presence of metal fragments in the abomasum, consistent with copper oxide wire particles. Although this disease is less common in goats, this case indicates that copper toxicity should be on the differential diagnostic list in goats with liver injury or an oxidative hemolytic anemia.
References
- Smith, M. and Sherman, D. ed., 2009. Blood, Lymph, and Immune System. In: Goat Medicine, 2nd ed. Wiley-Blackwell, pp.302-305.
- Hinchcliff, K., Lavoie, J. and Brown, C. ed., 2008. Anemia, Heinz Body. In: Blackwell’s Five-Minute Veterinary Consult: Equine. Wiley-Blackwell, pp.68-69.
- Stockham, S. and Scott, M. ed., 2008. Erythrocytes. In: Fundamentals of Veterinary Clinical Pathology, 2nd ed. Blackwell Publishing, pp.157-158.
- Gupta, R., 2018. A Review of Copper Poisoning in Animals: Sheep, Goat and Cattle. International Journal of Veterinary Sciences and Animal Husbandry, [online] 3(5), pp.1-4. Available at: <https://www.veterinarypaper.com/pdf/2018/vol3issue5/PartA/3-4-16-920.pdf>
- Bozynski, C., Evans, T., Kim Dae, Y., Johnson, G., Hughes-Hanks, J., Mitchell, W., Rottinghaus, G., Perry, J. and Middleton, J., 2009. Copper Toxicosis with Hemolysis and Hemoglobinuric Nephrosis in Three Adult Boer Goats. Journal of Veterinary Diagnostic Investigation, [online] 21(3), pp.395–400. Available at: <https://pubmed.ncbi.nlm.nih.gov/19407099/>
- Blakley, B., 2013. Overview Of Copper Poisoning. [online] Merck Veterinary Manual. Available at: <https://www.merckvetmanual.com/toxicology/copper-poisoning/overview-of-copper-poisoning>
- Constable, P., Hinchcliff, K., Done, S. and Grunberg, W. ed., 2017. Toxins Affecting the Hemolymphatic System. In: Veterinary Medicine: A Textbook of the Diseases of Cattle, Horses, Sheep, Pigs and Goats, 11th ed. Elsevier, pp. 823.
- DVM 360. n.d. Copper Toxicity In Sheep And Goats. [online] Available at: <https://www.dvm360.com/view/copper-toxicity-sheep-and-goats-proceedings>
Authors: E. Held (intern) and T. Stokol