
Interpretation
Neutrophilic inflammation with a protozoal infection; toxoplasmosis presumptive
Explanation
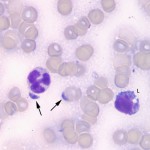
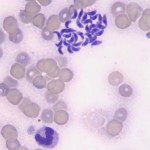
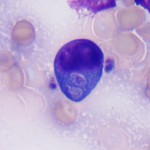
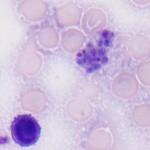
The lymph node aspirate was hemodiluted, with a moderate sampling of lymph node constituents. The lymphocytes were a mixed population of mostly small cells, with increased proportions of large lymphocytes (not shown). Some of the small and large lymphocytes had red cytoplasmic granules (not shown). There were also increased numbers of non-degenerate (but toxic) neutrophils (Figure 2), with a few vacuolated macrophages. Moderate numbers of crescentic to oval protozoal organisms (zoites) were present in the background (Figures 2 and 3), with some being observed ithin neutrophils, macrophages and large lymphocytes (Figure 4). A few platelet clumps mimicked the organisms (Figure 5). No other infectious agents were seen. The cytologic diagnosis was lymphoid hyperplasia with concurrent suppurative inflammation and a protozoal infection. The differential diagnoses for the organism were Toxoplasma, Neospora and Sarcocystis sp (Question 1).
Tissue inflammation or necrosis incited by the organism would explain the systemic inflammatory response (inflammatory leukogram, hypoalbuminemia). The borderline thrombocytopenia is concerning for a consumptive process (disseminated intravascular coagulation or DIC), secondary to inflammation. The high ALT and AST support liver injury, although both of these enzymes could be reflecting muscle injury, since the CK is high (and the AST is increased to a greater extent than the ALT). The decreases in electrolytes can be attributed to diarrhea and anorexia (low potassium) with a likely concurrent dilutional component from ADH (stimulated by hypovolemia). Concurrent losses of albumin into the gatrointestinal tract could be contributing to the hypoalbuminemia. The high urea nitrogen with normal creatinine could indicate a prerenal azotemia (from fluid losses in diarrhea) or early renal azotemia (a urinalysis was not performed). Concurrent stress (endogenous corticosteroid release) or an epinephrine response is likely responsible for the hyperglycemia (Question 2). Note, that the clinical pathologic results were interpreted using the mean ± 2xSD from ISIS values from male red-necked wallabies > 1 year of age. This assumes that the laboratory data is parametric, which is seldom the case with most clinical pathologic tests. However, since reference intervals are usually not available for exotic animals, published data should be used as rough guides, along with clinical judgement, in interpreting laboratory changes (i.e. even though the number of band neutrophils was within reference intervals, the presence of toxic change supported an inflammatory leukogram).
|
|
|
|
|
Further information and follow up
The wallaby was treated with subcutaneous fluids, antibiotics (enrofloxacin and metronidazole), an anti-foaming agent to reduce bloat (simethicone), and a non-steroidal anti-inflammatory agent (meloxicam), with assist feeding. Slight improvement was seen, although the diarrhea persisted. Based on cytologic evidence of a protozoal infection, a modified agglutination test (MAT) was performed on the wallaby’s serum for IgG antibodies against Toxoplasma gondii, the most likely pathogen (Question 3). Paired serum samples are usually recommended to confirm acute infection and reduce false negative test results in early infection. While only one sample was submitted, results for the wallaby were consistent with T. gondii infection (titer of 54,000; result >60 suggestive of acute or chronic infection). Two days later, the wallaby became depressed and lethargic, began excessively grinding his teeth and developed abdominal distension. Repeat abdominal radiographs and ultrasound revealed a severely gas-distended stomach, with a non-motile pyloric region, and intestines. No obvious obstruction was seen. A moderate amount of abdominal fluid was noted, which was aspirated and submitted for cytologic examination. This revealed a transudative effusion, with no evidence of organisms. A repeat hemogram revealed worsening inflammation (more band neutrophils and more severe toxic change), with a moderate thrombocytopenia (44 x 103/μL), and evidence of erythrocyte fragmentation injury (keratocytes). This supports DIC, likely as a consequence of tissue necrosis and inflammation (a coagulation panel was not performed to confirm this). A repeat biochemistry profile (see Table below) revealed worsening azotemia (renal, presumptive) and ongoing electrolyte losses (severe hyponatremia; secondary to ileus and continued diarrhea) and muscle injury (increases in CK and AST). The wallaby had a stress hyperglycemia and a hyperchloremic metabolic acidosis. Based on the histologic evidence of pulmonary edema and a mixed bacterial and protozoal pneumonia (see below), the acidosis could be a compensatory response to a primary respiratory alkalosis, but considering the defects in renal function, a primary bicarbonate loss acidosis due to the diarrhea was suspected (a blood gas analysis was not performed on the wallaby). Hyperglobulinemia indicated an inflammatory response or antigenic stimulation. A stomach tube was passed to temporally relieve distension and the wallaby was taken to surgery for an exploratory laparotomy. Unfortunately, the wallaby went into cardiac arrest and could not be resuscitated during surgery.
| Test | Day 1 | Day 3 |
| Sodium (mEq/L) | 130 | 125 |
| Potassium (mEq/L) | 2.2 | 3.3 |
| Chloride (mEq/L) | 85 | 85 |
| Bicarbonate (mEq/L) | 32 | 20 |
| Urea nitrogen (mg/dL) | 55 | 141 |
| Creatinine (mg/dL) | 1.5 | 3.8 |
| Albumin (g/dL) | 2.3 | 2.4 |
| Globulin (g/dL) | 3.4 | 4.1 |
| Glucose (mg/dL) | 218 | 433 |
| ALT (U/L) | 491 | 365 |
| AST (U/L) | 898 | 1356 |
| ALP (U/L) | 1048 | 1400 |
| GGT (U/L) | 27 | 28 |
| Total bilirubin (mg/dL) | 0.3 | 0.4 |
| Creatine kinase (U/L) | 25039 | 76551 |
Discussion
Toxoplasma gondii is a protozoan parasite, the only known definitive host of which is the cat. Oocysts are shed in cat feces however they are not infective until sporulation, which occurs 1-5 days after shedding. Once infective, sporulated oocysts can infect a wide variety of warm-blooded animals, including humans. Following ingestion of sporulated oocysts, sporozoites are released in the intestine, invade intestinal epithelium and local lymph nodes, and convert to tachyzoites. Tachyzoites are then able to spread hematogenously to all tissues, where they continue to proliferate. Following widespread proliferation, tachyzoites convert to bradyzoites, which are encysted within the tissues for the lifetime of the host. The life cycle is completed when a cat ingests a paratenic host harboring bradyzoites, allowing the protozoan to undergo both asexual and sexual reproduction, ending in the production and shedding of further oocysts.1
In warm-blooded animals, toxoplasmosis can manifest with a variety of clinical signs. It is a common cause of death in marsupials, particularly wallabies, in which it can manifest as sudden death with no premonitory signs, or with overt clinical disease.2-6 The increased susceptibility of marsupials to toxoplasmosis has been attributed to a lack of historical exposure, as felids were not present in Australia until the arrival of Europeans 300 years ago.2 As with other paratenic hosts, the most common source of infection is ingestion of sporulated oocysts from the environment (from the household or other cats). Overt toxoplasmosis may be seen due to acute infection or spontaneous recrudescence of disease. The most common clinical signs are respiratory and neurologic in nature, with dyspnea, tachypnea, incoordination, tremors, paresis, and blindness. Lethargy, diarrhea, and inappetence are also reported.3
Diagnosis of a protozoal infection antemortem can be achieved via cytologic examination of aspirates of infected tissues, but cytology cannot differentiate between different protozoa. On rare occasions, organisms may be seen in leukocytes (neutrophils and monocytes) in peripheral blood.7 Speciation is performed by serology or PCR. Postmortem, the diagnosis is confirmed by the presence of cysts with characteristic necrotizing lesions in multiple tissues and positive immunohistochemical staining for the specific organism. Gross lesions include pulmonary congestion, edema, and consolidation, lymphadenopathy, gastrointestinal ulceration, and splenomegaly.2-3 Severe interstitial pneumonia with pulmonary congestion, edema, necrosis, fibrosis, and alveolar histiocytosis is commonly observed in lung tissue. In lymph nodes, germinal center necrosis, lymphoid depletion, edema, congestion, and hemorrhage are documented. Histologic changes in gastrointestinal tissue include necrosis, inflammation, lymphoid infiltration, and mucosal ulceration.3
Toxoplasmosis in wallabies has been successfully treated with atovaquone (hydroxynaphthoquinone), an antimicrobial agent active against tachyzoites and bradyzoites. Atovaquone has also been used to treat toxoplasmosis in AIDS patients, and it has a synergistic effect when combined with clindamycin and pyrimethamine.6 Interestingly, while multiple concurrent infections with T. gondii were previously thought to be rare due to strong immunity elicited during primary infection, a recent DNA sequencing study of 16 free-ranging macropods found that 14 were infected with several distinct T. gondii genotypes in different organs.8
References
- Bowman DB. Georgis’ parasitology for veterinarians. 2012. St. Louis, MO. Saunders Elsevier; pp 101-102.
- Reddacliff GL, Hartley WJ, et al. 1993. Pathology of experimentally-induced, acute toxoplasmosis in macropods. Australian Vet J. 1: 4-6.
- Canfield PJ, Hartley WJ, et al. 1990. Lesions of toxoplasmosis in Australian marsupials. J. Comp. Path. 103: 159-167.
- Hartley MP. 2006. Toxoplasma gondii infection in two common wombats (Vombatus ursinus). Australian Vet J. 84: 107-109.
- Bermudez R, Failde LD, et al. 2009. Toxoplasmosis in Bennett’s wallabies (Macropus rufogriseus) in Spain. Vet Parasit. 160: 155-1587.
- Dubey JP, Crutchley C. 2008. Toxoplasmosis in wallabies (Macropus rufogriseus and Macropus eugenii): blindness, treatment with atovaquone, and isolation of Toxoplasma gondii. J of Parasit. 94: 929-933.
- Adkesson MJ, Gorman ME, Hsiao V, Whittington JK, Langan JN. 2008. Toxoplasma gondii inclusions in peripheral blood leukocytes of a red-necked wallaby (Macropus rufogriseus). Vet Clin Pathol 36: 97-100.
- Pan S, Thompson RC, et al. 2012. Western Australian marsupials are multiply infected with genetically diverse strains of Toxoplasma gondii. PLOS ONE 7: 1-6.