
The body tries to minimize pH changes and responds to acid-base disturbances with body buffers, compensatory responses by the lungs and kidney (to metabolic and respiratory disturbances, respectively) and by the kidney attempting to correct for a metabolic disturbances.
- Available body buffers: There are intracellular and extracellular buffers for primary respiratory and metabolic acid-base disturbances. Extracellular buffers are carbonate (HCO3–) and non-carbonate (e.g. protein in plasma) buffers. These immediately buffer the rise or fall in H+, however carbonate buffers cannot buffer pCO2 or respiratory changes. Intracellular buffers include hemoglobin in erythrocytes and phosphates in all cells – buffering with these take longer (several hours).
- Compensatory responses: This involves responses by the respiratory tract and kidney to primary metabolic and respiratory acid-base disturbances, respectively. Compensation is driven by pH changes and opposes the primary disturbance, although the laboratory changes in the compensatory response parallel those in the primary response. This concept is illustrated in the summary below.
- Respiratory compensation for a primary metabolic disturbance: Alterations in alveolar ventilation occurs in response to primary metabolic acid-base disturbances because the body senses changes in pH with peripheral chemoreceptors. This begins within minutes to hours of an acute primary metabolic disturbance. Note that complete compensation via this mechanism takes up to 24 hours.
- Renal compensation for a primary respiratory disturbance: Here, the kidney alters excretion of acid (excrete an acid, gain a base; retain an acid, excrete a base) in response to primary respiratory disturbances. This begins within hours of an acute respiratory disturbance, but take several days (3-5 days) to take full effect.
- Correction of acid-base changes: Correction of a primary respiratory acid-base abnormality usually requires medical or surgical intervention of the primary problem causing the acid-base disturbance, e.g. surgical relief of a collapsed trachea that is causing a primary respiratory acidosis (this is because the lungs cannot correct themselves, but need help to do so). Once the collapsed trachea is resolved, the lungs can rapidly blow off accumulated carbon dioxide. The kidney plays a major role in attempting to correct metabolic disturbances, through alterations of H+ or HCO3– excretion or absorption. This requires a normal kidney that is functioning appropriately. However, mild renal dysfunction will not affect the ability of the kidney to respond to acid-base disturbances. It requires fairly decent renal impairment (of tubular function, primarily) with acute kidney injury or chronic renal disease to result in defects in acid-base responses. For more information on how the kidney modulates acid base status (including diagrams), refer to the renal physiology page. In the case of metabolic alkalosis, the kidney’s ability to correct the alkalosis can be impaired and the kidney can actually perpetuate or worsen the alkalosis by excreting acid (called a paradoxic aciduria; see more below). Thus, correction of a primary metabolic disturbance also requires surgical or medical intervention, unless the primary disturbance is mild or self-limiting.
Metabolic disturbances
In primary metabolic acid-base disorders, the body responds initially through plasma and interstitial or extracellular buffers. Bicarbonate is the dominant buffer in a primary metabolic acidosis. It takes a little longer (hours) for intracellular buffering with hydrogen shifts on proteins to occur, but this starts to happen as well. We would also expect to find immediate compensatory changes in alveolar ventilation and pCO2 (i.e. secondary respiratory compensation). With time, the kidney should act to correct the metabolic disturbance, particularly after we fix the primary problem or if the primary problem was self-limiting or mild. There is some evidence that cats cannot correct a primary metabolic disturbance like dogs can (Di Bartola). Note, respiratory compensation will not return pH to normal in a primary metabolic acidosis. This requires cessation of the originating cause and medical or surgical intervention, as required, followed by renal correction.
The following can be used as a guide to estimate the expected respiratory compensatory response to primary metabolic acid-base disturbances in dogs (it is unclear if this formula works for other species, but it is all we have):
pCO2 (expected) = pCO2 (normal) + ([HCO3–(measured) – HCO3–(normal)] x 0.7) ± X
where, X=3 for a metabolic acidosis or X=2 for a metabolic alkalosis. Note that some authors use a correction factor of 1.2 versus 0.7.
Alternatively, the maximum respiratory compensation expected in a primary metabolic acidosis in dogs is:
pCO2 (expected) = 1.5 [HCO3–] + 8.
Response to a primary metabolic acidosis
The body’s response to a primary metabolic acidosis consists of body buffers and a compensatory respiratory alkalosis. Renal acid excretion (ammoniagenesis, vacuolar-type H+-ATPase activity) serves to attempt to correct the acidosis, but requires time (days) and a functioning kidney, as well as cessation of the primary problem (and respiratory compensatory response). There are also species differences in the body’s response to a metabolic acidosis. As indicated above, the kidney in cats, in general, are not as good as responding to the acidosis as well as dogs.
- Body buffers: In an acute primary metabolic acidosis, the acid load (high H+) is buffered by extracellular bicarbonate in plasma and interstitial fluid (around 40%). The plasma bicarbonate concentration thus decreases. There are also plasma non-carbonate buffers, such as plasma proteins, but these buffer around 1% of the acid load. As the hydrogen moves from the extracellular to the interstitial space and then intracellularly, intracellular buffers such as protein and phosphates in soft tissues and bone and hemoglobin in red blood cells will bind the excess hydrogen. The hydrogen moves in exchange for Na+ or K+ or, in the case of red blood cells, the H+ enters with chloride (these exchanges maintain electroneutrality), when HCl is given to induce an acidosis. Bone is also a good sink for hydrogen, exchanging for calcium (which can cause osteolysis in a chronic metabolic acidosis). Intracellular buffers thus are a sink for most of the acid load (50% or more).
- Respiratory compensation (alkalosis): A primary metabolic acidosis stimulates peripheral chemoreceptors (which respond to low pH or high H+), causing hyperventilation and a decrease in pCO2 or a secondary respiratory alkalosis. Hyperventilation is due to an increase in tidal volume rather than increased respiratory rate and, if severe, results in deep breaths, called Kussmaul’s respiration (which is a good clinical indicator of an underlying primary metabolic acidosis).
- Correction: The kidney attempts to correct for a primary metabolic acidosis by excreting excess acid as ammonium chloride, usually through enhanced renal ammoniagenesis by the proximal convoluted tubules of the kidney (see image below and renal physiology page). This regenerates the bicarbonate being consumed in plasma and creates a medullary interstitium that is high in ammonia, in fact 3 bicarbonate molecules are generated for each ammonia produced in the renal tubules from glutamate. This response takes several days to take effect and will correct the acidosis (and pH change), if the primary disturbance has ceased and given time and normal renal function (this does not appear to work as well in cats as in dogs and other species are an unknown commodity). The pH is not expected to normalize with an attempted corrective response if the primary metabolic acidosis is still ongoing and driving the pH down. The kidney can also excrete hydrogen actively in the collecting tubules via the V-H-ATPase. In the distal nephron, the hydrogen combines with ammonia (NH3) which passively diffuses across the cell from the interstitium into the tubular lumen. Once NH4+ is formed, it cannot diffuse back across the membrane and is stuck in the fluid, so it is excreted with chloride, that is still in the urine lumen or that has entered the urine lumen from passively following the hydrogen. So even though ammoniagenesis occurs primarily in the proximal convoluted tubule, it also allows the distal nephron to excrete hydrogen (with chloride). Interestingly, the V-H-ATPase in the type A intercalated cells in the cortical and medullary collecting tubules are usually in vesicles in the cytosol. Decreases in pH (particularly within the cell) drives the delivery of the pump to the luminal membrane, where it then actively excretes hydrogen with chloride passively following. The energy for this pump is driven by a basolateral bicarbonate/chloride exchanger, which pumps chloride into the cell in exchange for bicarbonate. The bicarbonate then increases in plasma and helps buffer the acidosis. So the corrective response to a primary metabolic acidosis is a metabolic alkalosis (loss of a chloride-containing acid, either NH4Cl or NH3 + HCl).
- Unfortunately, this corrective response to a primary metabolic acidosis is often impaired because the kidney cannot sense that the secondary hypocapnea is a good compensatory response to the original metabolic acidosis. Instead, the increase in intracellular pH from the loss of carbon dioxide associated with a compensatory respiratory alkalosis (carbon dioxide readily diffuses across membranes within the renal tubules) also serves to enhance the acidosis, because the increased intracellular pH reduces ammoniagenesis and prevents movement of the V-H-ATPase to the luminal membrane (which would normally result in acid excretion, so you get acid retention instead). This so-called maladaptive response by the kidney to the compensatory respiratory alkalosis to the original acidosis actually increases the acid load (by around 40%) (Madias et al 1977). Note this only occurs in a chronic metabolic acidosis and not an acute metabolic acidosis.
Response to a primary metabolic alkalosis
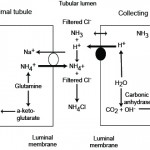
A primary metabolic alkalosis is initially offset by body buffers (not bicarbonate) and a compensatory respiratory acidosis. Increased renal excretion of bicarbonate (through increased filtration of the bicarbonate through the glomeruli and active excretion of bicarbonate by type B intercalated cells in the collecting tubules of the distal nephron) can rapidly correct the alkalosis, particularly when mild or self-limiting. This corrective response of the kidney to a primary metabolic alkalosis is frequently impaired, as a consequence of the primary disease process causing the alkalosis, which results in acid and not base excretion by the kidney, thus worsening the alkalosis.
- Body buffers: Intracellular (mostly proteins and phosphate, with a little due to hemoglobin and lactate) and extracellular (protein) buffers release protons and buffer about 32% and 1% of the increased bicarbonate, respectively. The rest stays in plasma and must be excreted by the kidney.
- Respiratory compensation (acidosis): An increased pH from the primary metabolic alkalosis will drive hypoventilation with an appropriate increase in pCO2 or secondary respiratory acidosis. Even though this will decrease oxygen delivery and the pO2, the hypoxemia is usually insufficient to stimulate ventilation and impair the compensatory response. However, the respiratory compensation leads to a maladaptive response, in that the increased carbon dioxide readily diffuses intracellularly, combines with water to form carbonic acid, which dissociates into hydrogen and bicarbonate. The bicarbonate shifts out of the cell in exchange for chloride, which moves intracellularly. This so-called chloride shift increases the bicarbonate concentration in blood versus decreasing it. In dogs, where the respiratory compensation was prevented by hypoxia, the bicarbonate concentration was lower than with the compensatory response. In addition, the decreased intracellular pH in the renal tubules will promote excretion of hydrogen (and bicarbonate retention) through stimulating renal ammoniagenesis and the V-H-ATPase. The respiratory compensatory response is thought to contribute up to 40% of the bicarbonate load in a chronic metabolic alkalosis and is considered maladaptive (Madias et al 1980).
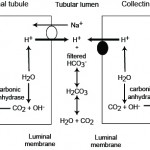
- Correction: The kidneys will attempt to correct for a primary metabolic alkalosis. More bicarbonate is filtered through the kidneys in a metabolic alkalosis. Also, a subpopulation of intercalated cells (type B) in the cortical collecting tubules of the kidney will excrete the excess HCO3– in a metabolic alkalosis. This is accomplished by translocation of the basolateral (blood side) bicarbonate/chloride exchanger (pendrin) to the luminal surface so bicarbonate is pumped into the urine and chloride is taken up (hence the correction is a hyperchloremic metabolic acidosis). This correction is not expected to normalize the pH unless the primary disturbance was mild and self-limiting or we have intervened to fix it. However, in many cases of a primary metabolic alkalosis, the ability of the kidney to excrete excess HCO3– is hampered by other factors which perpetuate the alkalosis (including the compensatory respiratory acidosis).
These factors (which result in a paradoxic aciduria) are:
- Hypovolemia: Most processes that result in a primary metabolic alkalosis also cause fluid loss, producing a hypovolemia. Hypovolemia will stimulate the renin-angiotension-aldosterone system. Angiotensin II promotes hydrogen excretion and bicarbonate retention by stimulating sodium absorption (with filtered bicarbonate) in the proximal tubule (by stimulating the luminal sodium/hydrogen antiporter and basolateral sodium/bicarbonate transporter). Aldosterone secretion also promotes renal HCO3– resorption and acid excretion (H+ loss), since aldosterone promotes the activity of the sodium transporter in principal cells (absorbs sodium, creating a lumen negative potential, which facilitates hydrogen excretion by type A intercalated cells in the cortical collecting tubule), V-H+-ATPases (which excrete hydrogen into the renal tubular lumen) and the basolateral chloride/bicarbonate exchanger (which retains bicarbonate) in type A intercalated cells in the collecting tubules.
- Hypochloremia: This is one of the main drivers of maintaining a metabolic alkalosis. Chloride is frequently depleted in conditions causing a metabolic alkalosis, because these conditions usually lose hydrogen with chloride (e.g. vomiting or sequestration of HCl).
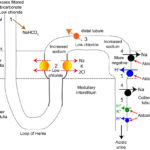
- In the PCT, sodium will be passively resorbed with filtered bicarbonate (which is in excess in blood and the glomerular filtrate) rather than chloride, which is depleted.
- Due to chloride depletion, more sodium is delivered to the distal nephron because less is being absorbed in the loop of Henle, which requires 2x chloride through the Na-K-2Cl carrier, and early distal tubule (NaCl transporter). Increased sodium delivery to the distal nephron will stimulate the Na transporter in the distal nephron (which is stimulated by aldosterone and the increased flow rate due to the excess sodium). This transporter absorbs the sodium in exchange for hydrogen (by stimulating the H+-ATPase in type A intercalated cells) or potassium, via potassium channels (which, if depleted, promotes aciduria in alkalosis, the very last thing you want to be happening).
- Chloride depletion will also decrease the activity of the Cl–/HCO3- pendrin cotransporter in type B and non-A and non-B intercalated cells in the collecting tubules, which can now no longer excrete the excess bicarbonate into urine.
- Decreased filtered chloride is a major stimulus for aldosterone secretion, because less NaCl is absorbed in the macula densa (reduced salt sensing). Aldosterone enhances sodium absorption in the collecting tubules by stimulating the Na transporter in principal cells at a time when more sodium is being delivered anyway because of decreased activity of the Na-K-2Cl and NaCl carriers in the loop of Henle and early distal tubules, respectively. Sodium exchanges for hydrogen, in cortical collecting tubular cells that have sodium dependent H+-ATPases, and potassium (H+/K+ transporters and other potassium channels, e.g. ROMK and maxi-H). If potassium is deficient, hydrogen moves out into the lumen instead, thus increasing renal acid excretion and, HCO3– retention. Aldosterone also directly promotes hydrogen excretion via stimulating the H+-ATPases in the collecting tubules.
Thus, the best treatment for a chloride-depleted metabolic alkalosis is to administer chloride as hypertonic NaCl (giving proportionally more chloride than sodium in relation to plasma in which sodium is higher than chloride), with supplemental potassium. This restores circulating volume (decreasing stimuli for angiotension II and aldosterone secretion), provides sodium (so less is absorbed in the proximal tubules with bicarbonate, allowing the bicarbonate to be filtered) and provides Cl–, allowing sodium to be resorbed with chloride in the distal parts of the proximal tubules and the loop of Henle, decreasing distal sodium delivery, and removing the stimulus for aldosterone secretion and acid excretion. Thus, NaCl is called an “acidifying solution”. Even though not acidic in itself, it promotes renal hydrogen retention and bicarbonate excretion (thus, it has an acidifying effect via the kidney).
- Hypokalemia: Potassium is often lost in fluids concurrently with H+ and Cl–. Potassium is also low because animals (cattle) are anorectic or it has moved intracellularly in exchange for hydrogen in alkalemic states. Potassium depletion is considered a critical element in sustaining a metabolic alkalosis in humans (Gennari 2011) but the same may not be true in dogs, where sodium avidity appears to dominate in hypovolemic or sodium chloride-deplete states (Dibartola).
- A low potassium will exacerbate hypovolemia (inhibits the action of ADH preventing water from being fully absorbed) and decreases GFR (reducing the amount of bicarbonate being filtered).
- Low potassium promotes bicarbonate resorption by stimulating the Na/K pump which transports bicarbonate out of the basolateral surface of renal tubular cells back into blood in the proximal tubule and stimulates renal ammoniagenesis in the proximal tubules (promoting ammonium chloride or acid excretion while retaining bicarbonate).
- Low potassium increases sodium delivery to the distal nephron by decreasing activity of the Na-K-2Cl carrier in the loop of Henle. The extra sodium delivered to the distal tubule is resorbed in exchange for H+ because K+ is low. In addition, the sodium absorption drives additional potassium excretion (even if deficient) by creating a negative lumen potential (just as it drives hydrogen excretion in cortical collecting tubule cells). Since aldosterone is high, sodium is being resorbed in the collecting tubules, but the low potassium causes hydrogen to move out instead of potassium.
Thus, a low K+ will potentiate an alkalosis by promoting renal acid excretion (and bicarbonate retention) in several ways.
Respiratory disturbances
The body’s response to primary respiratory acid-base disorders involves intracellular buffers followed by altered renal excretion of acid. There is little extracellular buffering capability for carbon dioxide (i.e. carbon dioxide moves rapidly across cell membranes and bicarbonate cannot buffer increases in carbon dioxide). Because of this, the body cannot offset an acute respiratory acidosis as well as a more chronic respiratory acidosis, where renal compensatory mechanisms kick in. This is why the factor in the equation below (Y) is low for acute and higher for chronic. Note, that the respiratory system cannot correct itself, i.e. the lungs cannot correct for a primary respiratory disturbance (unlike the kidney, which can correct a primary metabolic disturbance), unless the primary disturbance was mild and self-limiting (e.g. an anticipatory respiratory alkalosis occurs in sled dogs before a race due to excitement-induced hyperventilation). Usually, the veterinarian has to intervene to help the lungs correct a primary respiratory acid-base abnormality.
The following formulae can be used as guides to determine the expected metabolic compensatory responses to primary respiratory acid-base disorders:
HCO3– (expected) = HCO3–(normal) + ([pCO2(measured) – pCO2 (normal)] x Y) ± 2
where,
Y = 0.15 for an acute respiratory acidosis (< 24 hours)
Y = 0.35 for a chronic respiratory acidosis (> 48 hours)
Y = 0.25 for an acute respiratory alkalosis
Y = 0.55 for a chronic respiratory alkalosis
Response to a primary respiratory acidosis
A primary respiratory acidosis is initially offset by intracellular buffers, followed by increased renal acid excretion.
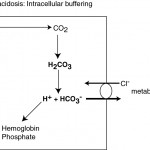
- Intracellular buffers: There is virtually no extracellular buffering capacity for increases in CO2 (bicarbonate cannot buffer itself). In an acute respiratory acidosis, CO2 moves rapidly intracellularly (particularly in red blood cells), where it disassociates with water to form H+ and HCO3–. The H+ is rapidly buffered by hemoglobin and intracellular phosphate. In red blood cells, the HCO3– moves extracellularly, in exchange for Cl–, causing an initial (usually mild) increase in plasma bicarbonate (metabolic alkalosis) and corrected hypochloremia (called the chloride shift).
- Metabolic compensation (alkalosis): The body compensates for the decreased pH from the primary respiratory acidosis by increasing renal excretion of H+. In a chronic respiratory acidosis, the body promotes acid excretion through the kidneys (through renal ammoniagenesis in the proximal convoluted tubules; see image above), which enhances HCO3– retention (3 x bicarbonate is retained for a single ammonium chloride that is excreted), i.e. the renal compensatory response is a secondary metabolic alkalosis. The kidneys can also directly excrete hydrogen through hydrogen pumps in the collecting tubules (type A intercalated cells), by stimulating movement of the V-H-ATPase to the luminal membrane, which occurs due to the decreased intracellular pH. The intracellular pH decreases more readily with respiratory than metabolic acidosis because carbon dioxide is freely diffusible across membranes and moves intracellularly. The kidney’s response begins within several hours of the onset of a respiratory acidosis, but like all other renal responses, takes 3-5 days to be of maximal effectiveness. Cats do not appear to respond as well as dogs to a chronic respiratory acidosis because they cannot substantially increase renal ammoniagenesis (Di Bartola). Note that there is some evidence that renal compensation may return the pH to normal in chronic respiratory acidosis in dogs given sufficient time (> 30 days) (Di Bartola). Even though pH may be normal in a chronic respiratory acidosis, the pCO2 will still be high. In fact, the increase in systemic pH from the renal compensatory response may be detrimental, because of suppression of ventilation.
Response to a primary respiratory alkalosis
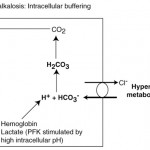
A primary respiratory alkalosis is initially offset by body buffers, followed by decreased renal acid excretion.
- Intracellular buffers: As for a primary respiratory acidosis, most of the initial buffering occurs intracellularly (release of H+ from protein, phosphate and hemoglobin buffers). Alkalemia also promotes intracellular lactic acid production, which also releases hydrogen. Chloride ions leave red blood cells in exchange for bicarbonate, causing a decrease in plasma bicarbonate (hyperchloremic metabolic acidosis) (see image to the right). This response is unlikely to affect the pH much as seen by the minor change in bicarbonate concentrations in an acute respiratory disturbance (see formula above).
- Metabolic compensation (hyperchloremic acidosis): In a chronic respiratory alkalosis, the increased pH will stimulate decreased renal acid excretion of H+ (decreased ammoniagenesis and lack of movement of V-H-ATPase to the luminal membrane in type A intercalated cells), with Cl– being retained to maintain electroneutrality. This response occurs within hours but takes several days for maximal effect. Thus, the compensatory response for a primary respiratory alkalosis is a hyperchloremic normal anion gap metabolic acidosis. In the dog, this may be so effective that the pH may return to normal if given sufficient time (14 days) (Di Bartola).
Compensation summary
- Compensation is opposite to the primary disturbance: metabolic compensation for a primary respiratory disturbance and respiratory compensation for a primary metabolic disturbance.
- Primary metabolic acidosis – secondary respiratory alkalosis
- Primary metabolic alkalosis – secondary respiratory acidosis
- Primary respiratory alkalosis – secondary metabolic acidosis (hyperchloremic, normal anion gap, acidosis)
- Primary respiratory acidosis – secondary metabolic alkalosis
- Compensatory laboratory changes parallel primary changes
- Primary metabolic acidosis = low HCO3-; compensatory respiratory alkalosis = low pCO2
- Primary metabolic alkalosis = high HCO3-; compensatory respiratory alkalosis = high pCO2
- Primary respiratory acidosis = high pCO2; compensatory metabolic alkalosis = high HCO3-
- Primary respiratory alkalosis = low pCO2; compensatory metabolic acidosis = low HCO3-
The table is a graphic representation of these concepts. As can be seen, the change in the primary parameter (HCO3– for metabolic and pCO2 for respiratory) is paralleled by the compensatory response.
| Primary disorder | pH | [H+] | Primary change | Compensation |
| Metabolic acidosis | ↓ | ↑ | ↓ HCO3– | ↓ pCO2 |
| Metabolic alkalosis | ↑ | ↓ | ↑ HCO3– | ↑ pCO2 |
| Respiratory acidosis | ↓ | ↑ | ↑ pCO2 | ↑ HCO3– |
| Respiratory alkalosis | ↑ | ↓ | ↓ pCO2 | ↓ HCO3– |
Related links
- Renal physiology: Information and diagrams on how the kidney excretes acid normally produced from daily metabolism (titratable acidity), reclaims bicarbonate normally filtered through the kidney (by absorbing filtered bicarbonate with sodium) and generates new bicarbonate while excreting excess acid (attempted correction for a primary metabolic acidosis not due to renal dysfunction or compensation for a primary respiratory alkalosis) through renal ammoniagenesis in the proximal convoluted tubules and excretion of hydrogen by hydrogen pumps (chloride passively follows) in the distal convoluted tubules.
References
- Clinical Physiology of Acid-Base and Electrolyte Disorders by Rose BD and Post DW, 5th edition, 2001. McGraw-Hill, New York, NY.
- Fluid, Electrolyte and Acid-Base Disorders in Small Animal Practice by DiBartola SP, 3rd edition, 2006. Elsevier-Saunders, St Louis, MO.
- Gennari 2011. Pathophysiology of Metabolic Alkalosis: A New Classification Based on the Centrality of Stimulated Collecting Duct Ion Transport. Am J Kidney Dis 58:626.