
Case Answer: Addison’s disease
The laboratory test results are consistent (in fact, these results are almost textbook!) for hypoadrenocorticism. Confirmatory testing is discussed in the follow-up section.
Explanation
CBC:
The complete blood count noticeably lacks a stress leukogram (specifically the lymphopenia) despite the animal’s emergent condition on presentation (Question 1). This may suggest a cortisol deficiency. The moderate leukocytosis and trending high eosinophils could be explained by a “reverse stress leukogram” as these cells are released into circulation at higher rates when there is no inhibitory action of cortisol.1
Electrolyte changes:
There is what appears to be a proportionate decline in Na and Cl. However, the dog has a history of vomiting (which is a common cause of selective Cl loss) so it is worthwhile to make sure the chloride “corrects” to the normal range. Note, the mid-range of the reference interval for Na can be used as the “normal Na value”
Corrected Cl– = (normal Na+/measured Na+) x measured Cl–
= (146/132.5) x 95
= 104.7
Talk about being just outside the reference interval (105-116 mEq/L). Despite the history of vomiting, the over-riding pattern is one of proportionate losses in Na and Cl. The two broad categories of mechanisms that lead to these electrolyte changes are loss of electrolyte-rich fluids (most commonly GI losses or third spacing) or retention of free water (ADH-driven in a state of perceived hypolvolemia; for instance, this is the underlying mechanism of fluid retention in cardiac failure). In this dog’s case, there is a more sinister mechanism and the K+ value helps elucidate this. Potassium can be increased for a number of reasons, including decreased renal excretion (post-renal obstruction or anuric renal failure) or a shift from intracellular stores to extracellular stores that occurs in cases of severe (mineral or hyperchloremic) acidosis (usually due to bicarbonate loss). In this case, the acidosis is likely the result of uremic acids and lactate (titration not loss), neither of which will cause an extracellular shift in K+. The animal is urinating, so a post-renal obstruction is not likely. This leads us to the hormone that facilitates both Na+ retention and K+ excretion: aldosterone.1,2 Aldosterone is a steroid hormone that works at the distal convoluted tubule of the nephron. In cases of low or absent aldosterone, Na+ is lost at an increased rate. Depletion of Na+ is followed by a loss of free water that can ultimately lead to a hypovolemic state and subsequent decreased tissue perfusion. Other differential diagnoses for the loss of Na+ include renal failure, gastrointestinal disease and loss, parasites (i.e. Trichuris), and pleural effusion. Some clinicians will look at the Na:K ratio to support their suspicions of hypoadrenocorticism, as a ratio less than 24:1 supports a diagnosis of Addison’s disease.4 However, this ratio can be normal in dogs that lack only glucocorticoids. The biochemical results from the dog in this case likely lacks both glucocorticoids(this is supported by the lack of stress leukogram and hypocholesterolemia) and mineralocorticoids.
Azotemia:
Azotemia can be pre-renal, renal, or post-renal in origin. Extra-renal factors, such as hypokalemia, hypercalcemia, plasma Na+ concentration, and bacterial lipoproteins (endotoxins), can also affect the ability of the renal tubule to function properly. The ability of the kidney to concentrate the urine requires a medullary concentration gradient (which relies on both urea and Na+), anti-diuretic hormone (ADH), and the ability of the tubules to respond to ADH. Dogs with Addison’s disease often have poorly concentrated urine due to extra-renal and pre-renal causes. The dog in this case has a high PCV, which supports dehydration (pre-renal). Also, if the Na+ has been chronically low, this removes the osmotic stimulus for ADH release.2 Hypercalcemia, which is present in this dog, can also interfere with concentrating ability by decreasing tubule responsiveness to ADH. Hypoxia, secondary to hypovolemia and a reduction in renal blood flow, may cause renal tubular damage. In this case, as is true with many Addisonian dogs, the azotemia resolved with fluid therapy (Question 3).
Hypocholesterolemia:
There are not many diseases that cause cholesterol to drop below the reference interval in the dog. Lack of synthesis due to liver failure or a portosystemic shunt, severe GI disease (protein-losing enteropathy), a paraneoplastic event (most often associated with multiple myeloma but we have projects underway at Cornell investigating this in a number of cancerous conditions), and lack of cortisol (Question 2). Why does a lack of cortisol lead to decreased plasma concentrations of cholesterol? The effect of glucocorticoids on cholesterol absorption, synthesis and excretion are complex and multifactorial. Cortisol mediates intestinal absorption of cholesterol, hence glucocorticoid-deficient animals do not absorb as much cholesterol as their steroid-replete counterparts.2 Glucocortocoids also impact cellular synthesis of cholesterol. This mechanism is more apparent in states of excess, which result in increased production of very low density lipoproteins by the liver.3 In culture, dexamethasone stimulates the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, a key enzyme in the synthesis of cholesterol, and both dexamethasone and cortisol stimulate cholesterol biosynthesis from human hepatocytes.4 Presumably, the low cholesterol levels seen in hypoadrenocorticism are also partially due to the loss of these stimulatory effects on production, although this has never been directly proven in the dog.
Acid base status:
The acid base status of Addison’s disease is often complex. The dog in this case has an acidemia (the blood pH is low). The high anion gap and low bicarbonate indicate a primary titrational metabolic acidosis. Given the dog’s hypovolemic state, lactic acid is a likely contributing factor as are phosphates and sulfates that are accumulating secondary to a decreased glomerular filtration rate (severe prerenal azotemia or renal azotemia). If the kidney is responding as it should to this acidosis (i.e. correction), one would expect the urine to have an acidic pH, as the result of H+ excretion by distal tubules. The normal pH of this dog’s urine suggests this is not occurring, which actually makes sense because aldosterone acts to increase excretion of H+ ions in the distal tubule. Aldosterone deficiency also causes increased potassium concentrations that then acts to decrease the amount of ammonium produced in the nephron. It has been suggested in the human literature that the tubular acidosis (from decreased excretion of hydrogen by the tubules) plays a role in the metabolic acidosis with canine Addison’s disease as well.6,10
Additional results and confirmatory testing
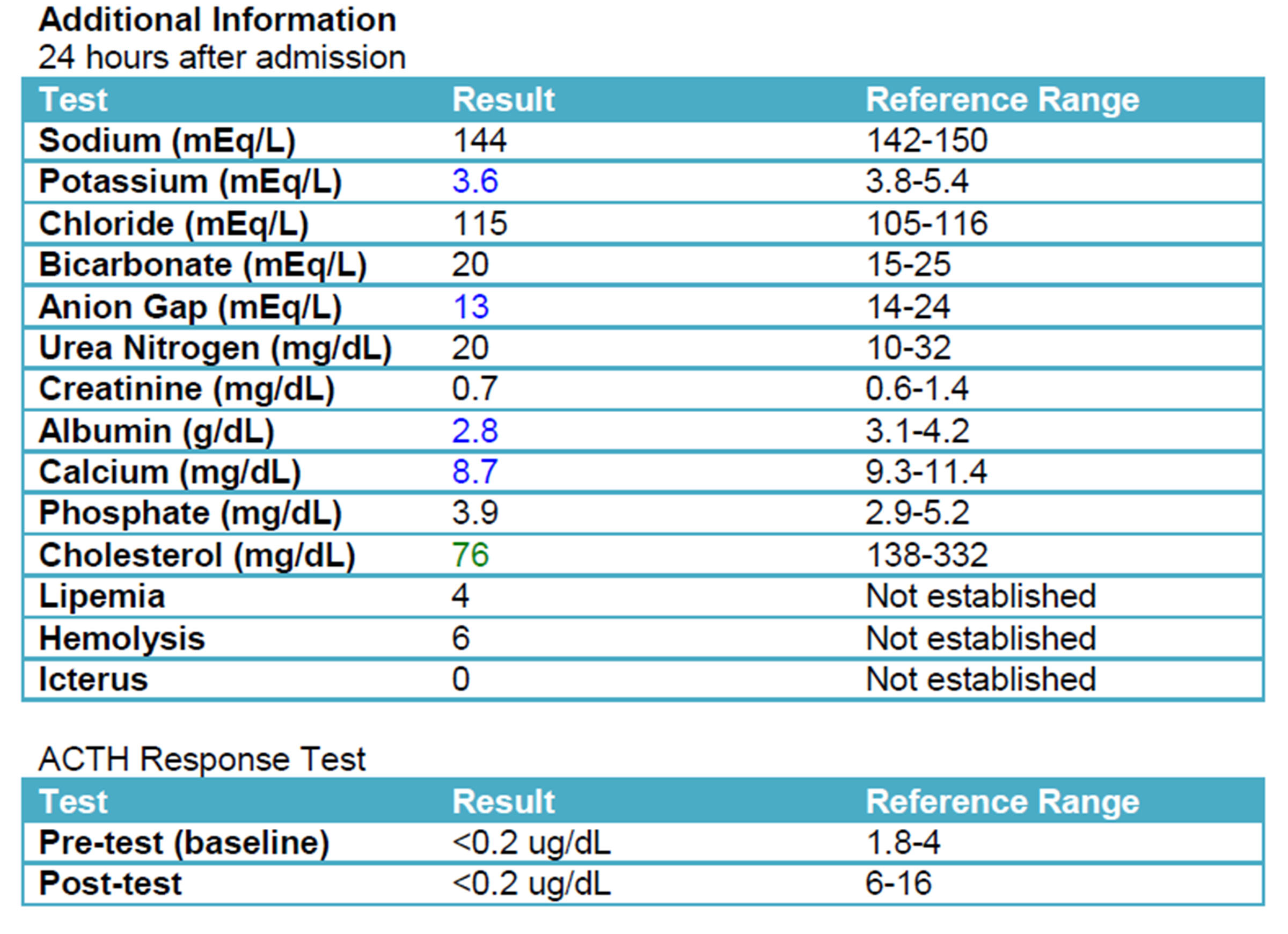
A repeat chemistry panel (see below) showed resolution of the azotemia and near normalization of electrolyte values. An ACTH stimulation test, which is considered the gold-standard for diagnosing hypoadrenocorticism, confirmed the suspected diagnosis.
Discussion
Primary hypoadrenocorticism, also known as Addison’s disease, is a condition in which dogs lack appropriate levels of glucocorticoids and mineralocorticoids. This is commonly suspected to be an immune-mediated process that leads to impairment of the adrenal cortex where glucocorticoids (primarily cortisol) and mineralocorticoids (primarily aldosterone) are produced. Deficiencies in these hormones have a wide range of systemic effects. This disease affects all breeds of dog, male or female, although the incidence in female dogs may be greater.10 Clinical signs are vague and Addison’s disease is known as “the great pretender”.1 The majority of dogs present with lethargy, weakness, inappetence, and gastrointestinal signs. Other common clinical signs include polyuria and polydipsia, and bradycardia.2, 12
A diagnosis of Addison’s disease can be made through clinical signs, physical examination findings, characteristic blood results, and an ACTH stimulation test. It is important to remember that not all dogs with Addison’s idsease, whether primary or secondary, will have characteristic electrolyte changes.12 An ACTH stimulation post-test result of less than 0.2 mg/dL, as was found in this patient, confirms the diagnosis of hypoadrenocorticism.13 Due to the nature of the electrolyte imbalances, an electrocardiogram could also be beneficial in work-up of a suspected Addisonian. An ECG would show increased amplitude of T waves, wide QRS complexes, and flattened R waves. These changes are a result of the high potassium. If the potassium continues to increase, the ECG will show bradycardia, flattening of the P-wave, followed by first-degree heart block.2,12 Arrhythmias such as sinoatrial standstill can also occur.9
As a result of its suspected immune-mediated nature, there is no curative treatment for Addison’s disease and dogs will likely need to remain on replacement therapy for the remainder of their lives.14 In a dog that presents as this patient did, stabilization with intravenous fluid administration should be implemented immediately. Ideally, this fluid should contain little to no potassium, as it is likely that this electrolyte is already increased. If the ECG is concerning, or blood-work reveals dangerously increased potassium, regular insulin or calcium gluconate should be given with the fluids.1,7 Hypoglycemia is a common finding in these dogs and dextrose should be added to the fluids to supplement them. At initial presentation, the dog should also receive a dose of glucocorticoids. This can be either prednisolone sodium succinate, or preferably dexamethasone, as prednisolone will cross-react with measurements of serum cortisol.14 Dogs with primary Addison’s disease will also need to be supplemented with mineralocorticoids, often in the form of oral fludrocortisone acetate or subcutaneous desoxycorticosterone pivalate (DOCP).15 So-called “atypical” Addisonian dogs are only deficient in glucocorticoids and therefore do not have theexpected electrolyte abnormalities. These cases will not require a mineralocorticoid as part of their treatment.12
Once the patient is stable and the crisis is resolved, glucocorticoid and mineralocorticoid supplementation can continue at physiologic doses. Frequent blood-work is recommended while titrating to ensure the lowest effective steroid dose, then every 4-6 months thereafter.13 In circumstances when the body would naturally elevate adrenal hormones, such as stressful situations, Addisonian dogs require higher doses of steroids to avoid a hypovolemic crisis state. Additional glucocorticoids are generally given prior to a known stressor at 2-5 times the maintenance dose.12 The prognosis for Addisonian dogs that are well controlled is good.5, 12
Follow-up
This patient was stabilized and treatment was initiated. Once the dog ate well on its own without vomiting, the dog was discharged. At recheck, 25 days after the start of treatment, the dog’s electrolytes remained well controlled and the patient was doing well clinically.
Historical note:

References
- Klein, S.C., and Peterson.. M. “Canine hypoadrenocorticism: part I.” The Canadian Veterinary Journal51.2. (2010): 179.
- Lathan P., and Tyler, J. “Canine hypoadrenocorticism: pathogenesis and clinical features.” Compendium on continuing education for the practicing veterinarian. (2005).
- McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol 2008;20(2):131-137.
- Carr BR, Simpson ER. Cholesterol synthesis by human fetal hepatocytes: effects of hormones. J Clin Endocrinol Metab. 1984 Jun;58(6):1111-6.
- Peterson M. et al. “Pretreatment clinical and laboratory findings in dogs with hypoadrenocorticism.” Journal of the American Veterinary Medicine Association. (1996): 85-91.
- Adler, Jennifer et al. “Abnormalities of serum electrolyte concentrations in dogs with hypoadrenocorticism.” (2007). Journal of veterinary internal medicine21.6 (2007): 1168-1173.
- Meeking, Susan. “Treatment of acute adrenal insufficiency.” (2007). Clinical techniques in small animal practice22.1: 36-39.
- Harvey, John et al. “Potential effects of glucocorticoids on serum iron concentration in dogs.” Veterinary Clinical Pathology2 (1987): 46-50.
- Adamama-Moraitou, K.K. et al. “Short-Term Exogenous Glucocorticosteroidal Effect on Iron and Copper Status in Canine Leishmaniasis (Leishmania Infantum).” Canadian Journal of Veterinary Research4 (2005): 287–292.
- Unwin, Robert J, and Giovambattista Capasso. “The Renal Tubular Acidoses.” (2001). Journal of the Royal Society of Medicine5: 221–225.
- Greco, Deborah S. “Hypoadrenocorticism in small animals.” Clinical techniques in small animal practice22.1 (2007): 32-35.
- Catharine J., and Scott-Moncrieff, R. “Hypoadrenocorticism.” In: Ettinger and Feldman’s Textbook of Veterinary Internal Medicine. 7th St. Louis, MO. Elsevier Saunders. (2010):847-1857.
- Bovens, C., et al. “Basal serum cortisol concentration as a screening test for hypoadrenocorticism in dogs.” Journal of Veterinary Internal Medicine5 (2014): 1541-1545.
- Klein, S.C., and Peterson, M. “Canine hypoadrenocorticism: part II.” The Canadian Veterinary Journal2 (2010): 179.
- Kintzer P., and Peterson, M. “Treatment and long term follow up of 205 dogs with hypoadrenocorticism.” Journal of Veterinary Internal Medicine.(1997):43-49.
Authorship: Ashley Bendlin (Class of 2017) and Dr. Erica Behling-Kelly