
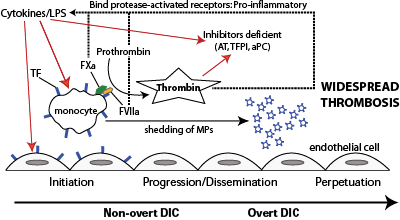
Inflammatory cytokines or lipopolysaccharide (LPS, endotoxin) stimulate tissue factor (TF) expression on monocytes and likely endothelial cells (initiation). TF binds to its enzymatic ligand, factor VII, which autoactivates. The TF-FVIIa complex then binds to and activates factor X, which generates thrombin. Thus, in bacterial sepsis, thrombin can be generated in the absence of endothelial injury (the physiologic stimulus for hemostasis activation). Thrombin amplifies its own production, activates and recruits platelets, and promotes clot formation, while inhibiting fibrinolysis. This is the non-overt or contained phase of DIC. Activated platelets and monocytes (and likely endothelial cells) shed procoagulant PS-bearing micro particles, which accelerate and disseminate thrombin formation. Inhibitors, such as antithrombin (AT), activated protein C (aPC) and tissue factor pathway inhibitor (TFPI) are downregulated as a direct consequence of the disease (inflammatory cytokines) or consumed (trying to inhibit the activated hemostatic system). Lack of inhibition facilitates dissemination and progression. Thrombin and activated coagulation factors (TF-FVIIa, TF-FVIIa-FXa) bind to protease activated receptors on cells (monocytes, endothelial cells) and incite a pro-inflammatory response, including cytokine release and up regulation of adhesion molecules. This perpetuates the inflammatory response, which positively feedbacks on the activated hemostatic system, setting up a vicious cycle that leads to overt DIC.