
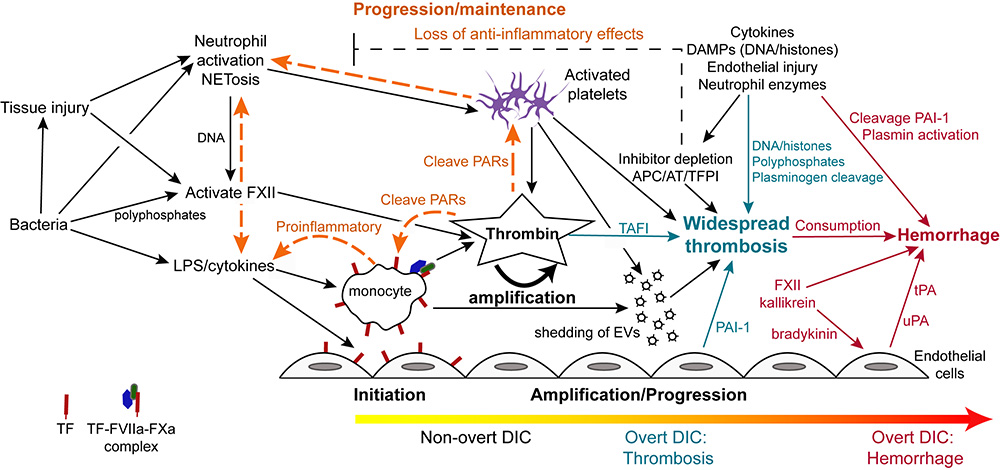
Disseminated intravascular coagulation is typically initiated by lipopolysaccharide (LPS)- or proinflammatory cytokine-induced upregulation of TF expression on monocytes and likely endothelial cells in sepsis due to gram negative bacteria. Tissue factor can also be expressed and released by other cells including platelets and eosinophils, but monocytes are thought to be the main source of TF in bacterial sepsis. In cancer, constitutive expression of TF on cancer cells and induction of TF on monocytes by proinflammatory cytokines is the likely trigger of DIC (not shown). Monocyte- or cancer-surface TF, FVIIa, and FXa form an active complex which generates thrombin. Bacteria, via release of long chain polyphosphates, or induction of tissue injury, inflammation, or NETosis, can also initiate DIC through activation of FXII. Bacteria can also activate platelets, promoting release of polyphosphates and procoagulant activity, via toll-like receptors (particularly TLR4, not shown). Endothelial injury, via release of long strings of von Willebrand factor, can capture and potentially activate circulating platelets in conditions associated with endothelial injury, providing a nidus for thrombin formation. Release of damage-associated molecular patterns (DAMPs, e.g. cell-free DNA) from injured tissues (including endothelial cells), inflammatory cells (via NETosis) or cancer can similarly trigger DIC via FXII and potentially other mechanisms (e.g. upregulation of TF by high-mobility group box protein 1, a nuclear associated inflammatory cytokine).
In early, non-overt DIC, inhibitors such as activated protein C (APC), antithrombin (AT) and tissue factor pathway inhibitor (TFPI), control the activated hemostatic system. If inhibitory mechanisms become overwhelmed, then thrombin amplifies its own production, simultaneously inhibiting fibrinolysis via thrombin-activatable fibrinolysis inhibitor (TAFI), resulting in microvascular thrombosis (progression/maintenance phase). This is now the hypercoagulable or thrombotic phase of overt DIC. Procoagulant extracellular vesicles (EVs) shed from thrombin-activated platelets and other cells, as well as DNA in extracellular traps, provide a negatively charged surface that supports systemic coagulation complex assembly, which is now longer held in check by inhibitors, which are rendered deficient or dysfunctional from DAMPs, pro-inflammatory cytokines and neutrophil proteases. Systemic thrombin production is also mediated by distant endothelial injury and dysfunction from microvascular thrombi, inflammatory mediators and histones. Inhibition of fibrinolysis via endothelial release of plasminogen activator inhibitor-1 (PAI-1), bacterial and platelet polyphosphates, DAMPs and neutrophil protease cleavage of plasminogen promotes persistence of thrombi. In the maintenance/pgroression phase, crosstalk between activated coagulation factors and platelets, such as through protease-activated receptor (PAR) signaling, potentiates the inflammatory response, including NETosis and release of proinflammatory cytokines from monocytes. The depletion of anti-inflammatory coagulation inhibitors further facilitates inflammation thereby maintaining the inflammation-DIC cycle. As coagulation factors are cleaved or consumed (along with platelets), DIC may shift to an overt hemorrhagic or hypocoagulable phase. This phase of DIC may also occur if profibrinolytic forces dominate, such as through PAI-1 cleavage PAI-1, FXII-, kallikrein-, protease-mediated cleavage of plasminogen to plasmin and release of tissue- (tPA; stimulated by bradykinin or endothelial injury) or urokinase-type plasminogen activators (uPA).